
Publications
Selected publications, 2025-26
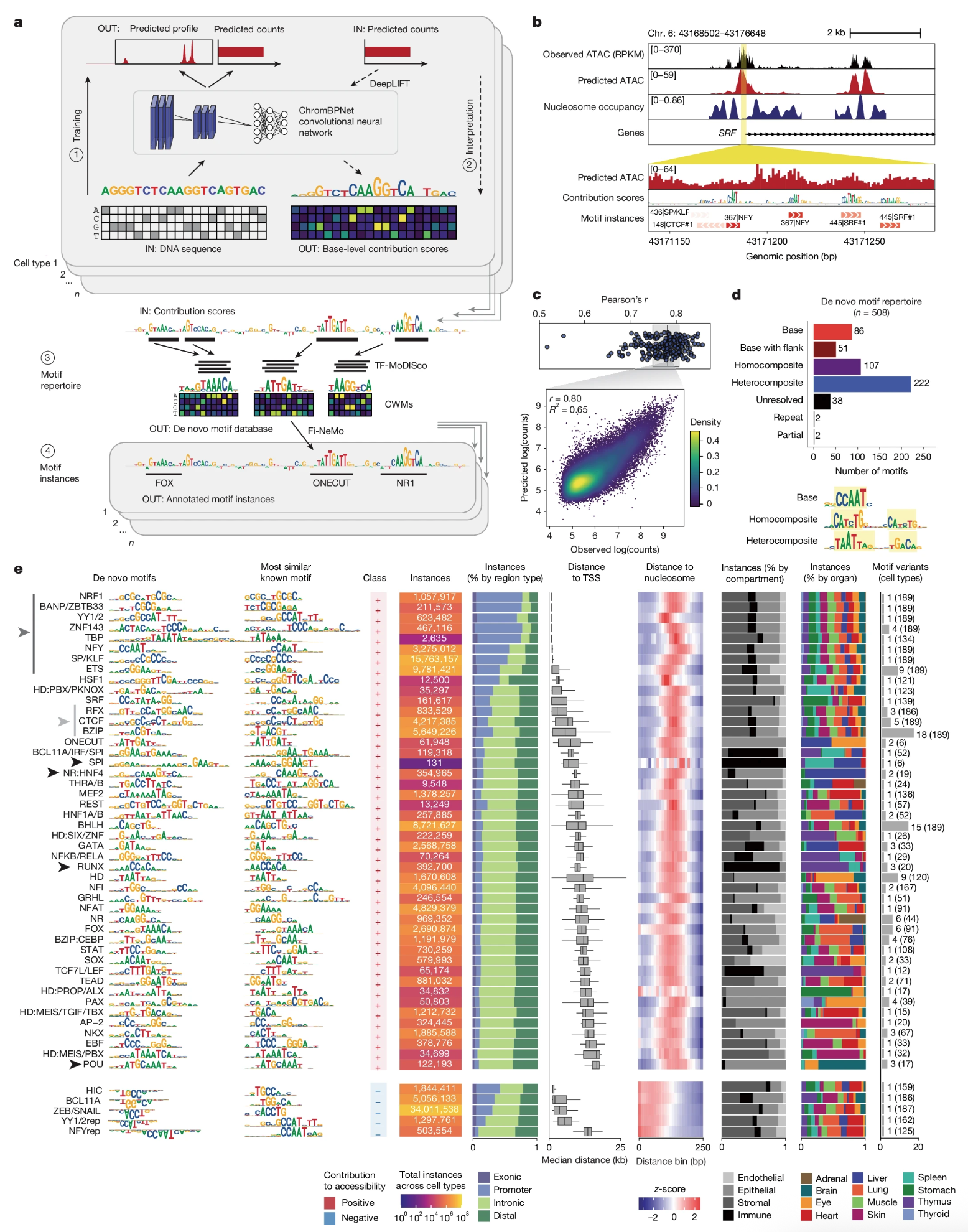
Betty B Liu, Selin Jessa, Samuel H Kim, Yan Ting Ng, Soon Il Higashino, Georgi K Marinov, Derek C Chen, Benjamin E Parks, Li Li, Tri C Nguyen, Austin T Wang, Sean K Wang, Meng How Tan, Serena Y Tan, Michael Kosicki, Len A Pennacchio, Eyal Ben-David, Anca M Pasca, Anshul Kundaje, Kyle KH Farh, William J Greenleaf (2026) Nature
Multiomics and deep learning dissect regulatory syntax in human development
Transcription factors establish cell identity during development by binding regulatory DNA in a sequence-specific manner, often promoting local chromatin accessibility and regulating gene expression1. Mapping accessible chromatin offers critical insights into transcriptional control, but available datasets for human development are restricted to bulk tissue, single organs or single modalities2. Here we present the Human Development Multiomic Atlas, a single-cell atlas of chromatin accessibility and gene expression from 817,740 fetal cells across 12 organs, spanning 203 cell types and more than 1 million candidate cis-regulatory elements, many of which exhibit organ-specific in vivo enhancer activity. Deep learning models trained to predict accessibility from local DNA sequence unravel a comprehensive lexicon of motifs that influence accessibility, including composite motifs exhibiting distinct syntactic constraints that are predicted to mediate transcription factor cooperativity. We identify ‘hard’ syntactic rules requiring precise motif spacing and orientation, ‘soft’ rules allowing flexible motif arrangements, and ubiquitous motifs inhibiting accessibility. Model-based interpretation of genetic variants reveals that disruption of motifs with positive and negative effects is associated with concordant effects on gene expression. Our work delineates how motif syntax governs cell-type-specific chromatin accessibility and provides a foundational resource for decoding cis-regulatory logic and interpreting genetic variation during human development.
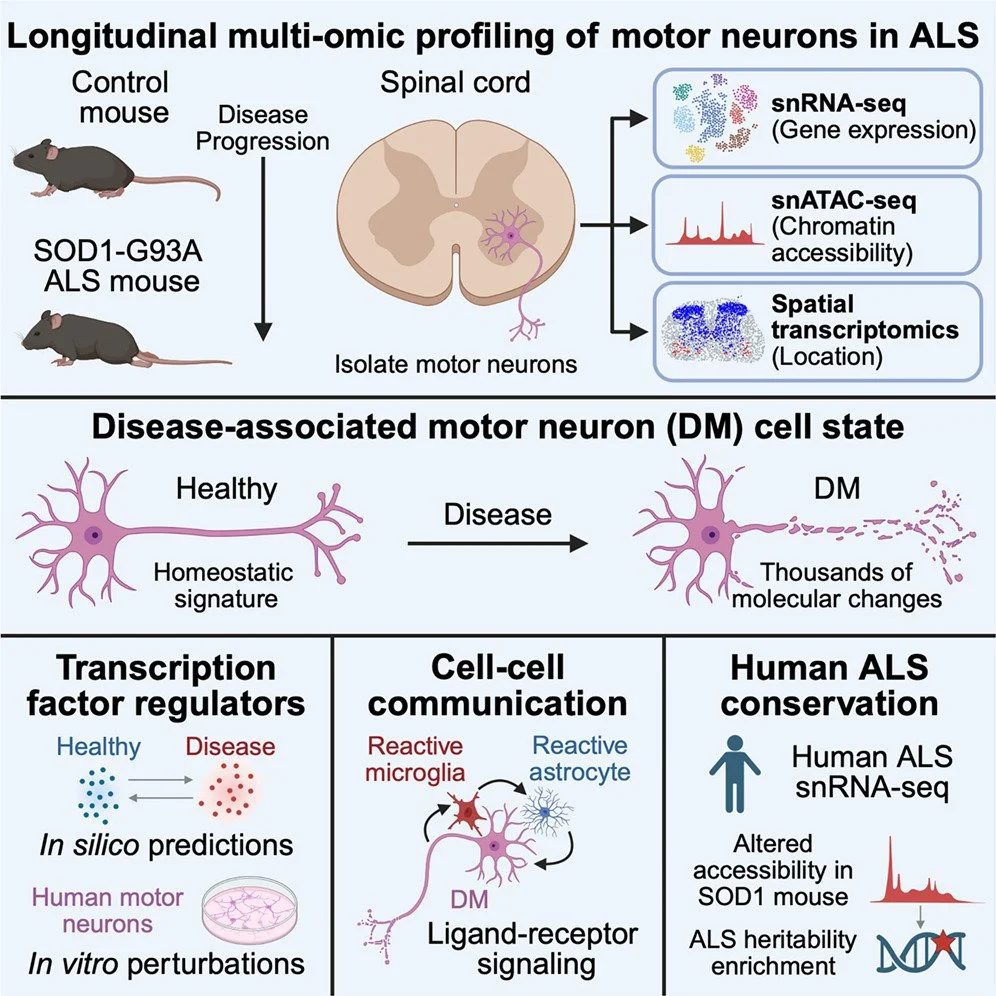
Olivia Gautier, Jacob A Blum, Thao P Nguyen, Shaolong Cao, Sandy Klemm, Mai Yamakawa, Dann Huh, Jessica A Hurt, Nasa Sinnott-Armstrong, Yi Zeng, Chung-ha O Davis, Juliane Bombosch, Chang Liu, Lisa N Encarnacion, Kevin A Guttenplan, Derek Chen, Arwa Kathiria, Luke Zhao, Stephen Moore, Alex Meng, Kailee Ong, Don W Cleveland, John Ravits, Jessica E Rexach, William J Greenleaf, Aaron D Gitler (2026) Cell
An emergent disease-associated motor neuron state precedes cell death in ALS
To define molecular determinants of motor neuron degeneration in amyotrophic lateral sclerosis (ALS), we generated longitudinal single-nucleus transcriptomes and chromatin accessibility profiles of spinal motor neurons together with spatial transcriptomics from the SOD1-G93A mouse model. Vulnerable alpha motor neurons showed thousands of molecular changes, marking a transition into a distinct cell state we named “disease-associated motor neurons” (DMs). We identified transcription factor networks that govern how healthy cells transition into DMs and those associated with motor neuron subtype-selective vulnerability. Upregulation of DM-associated transcription factors in human motor neurons induced key features of DMs, demonstrating an active regulatory component. Human ALS spinal cord single-nucleus RNA sequencing data demonstrated conservation of the DM signature in alpha motor neurons, and human orthologs of regions differentially accessible in SOD1-G93A mouse motor neurons were enriched for ALS genetic risk variants. Together, these findings establish a conserved, genetically linked motor neuron signature in ALS.
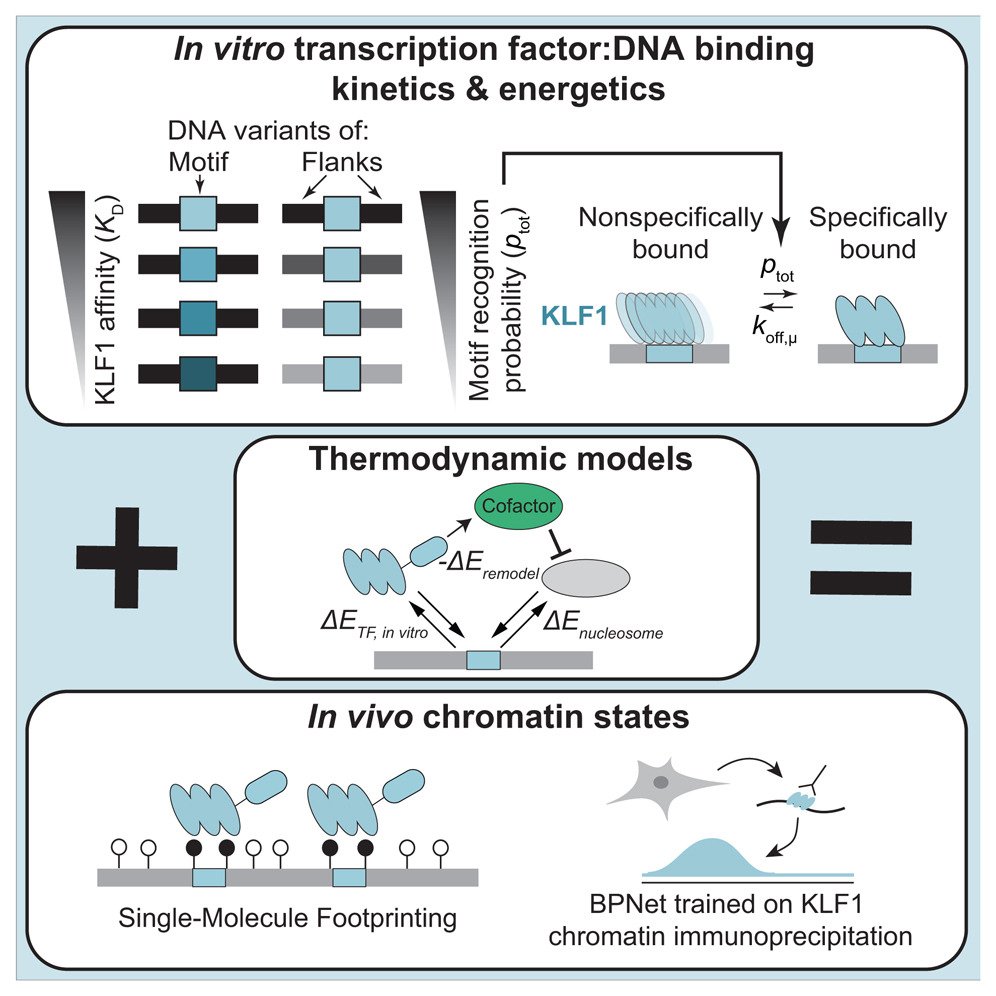
Julia M Schaepe, Torbjörn Fries, Benjamin R Doughty, Vivekanandan Ramalingam, Betty B Liu, Olivia J Crocker, Georgi K Marinov, Michaela M Hinks, Emil Marklund, William J Greenleaf (2025) Cell
Thermodynamic principles link in vitro transcription factor affinities to single-molecule chromatin states in cells
The molecular details governing transcription factor (TF) binding and the formation of accessible chromatin are not yet quantitatively understood—including how sequence context modulates affinity, how TFs search DNA, the kinetics of TF occupancy, and how motif grammars coordinate binding. To resolve these questions for a human TF, erythroid Krüppel-like factor (eKLF/KLF1), we quantitatively compare, in high throughput, in vitro TF binding rates and affinities with in vivo single-molecule TF and nucleosome occupancies and in vivo-derived deep learning models. We find that 40-fold flanking sequence effects on affinity are consistent with distal flanks tuning TF search parameters and captured by a linear energy model. Motif recognition probability, rather than time in the bound state, drives affinity changes, and in vitro and in nuclei measurements exhibit consistent, minutes-long TF residence times. Finally, in vitro biophysical parameters predict in vivo sequence preferences and single-molecule chromatin states for unseen motif grammars.
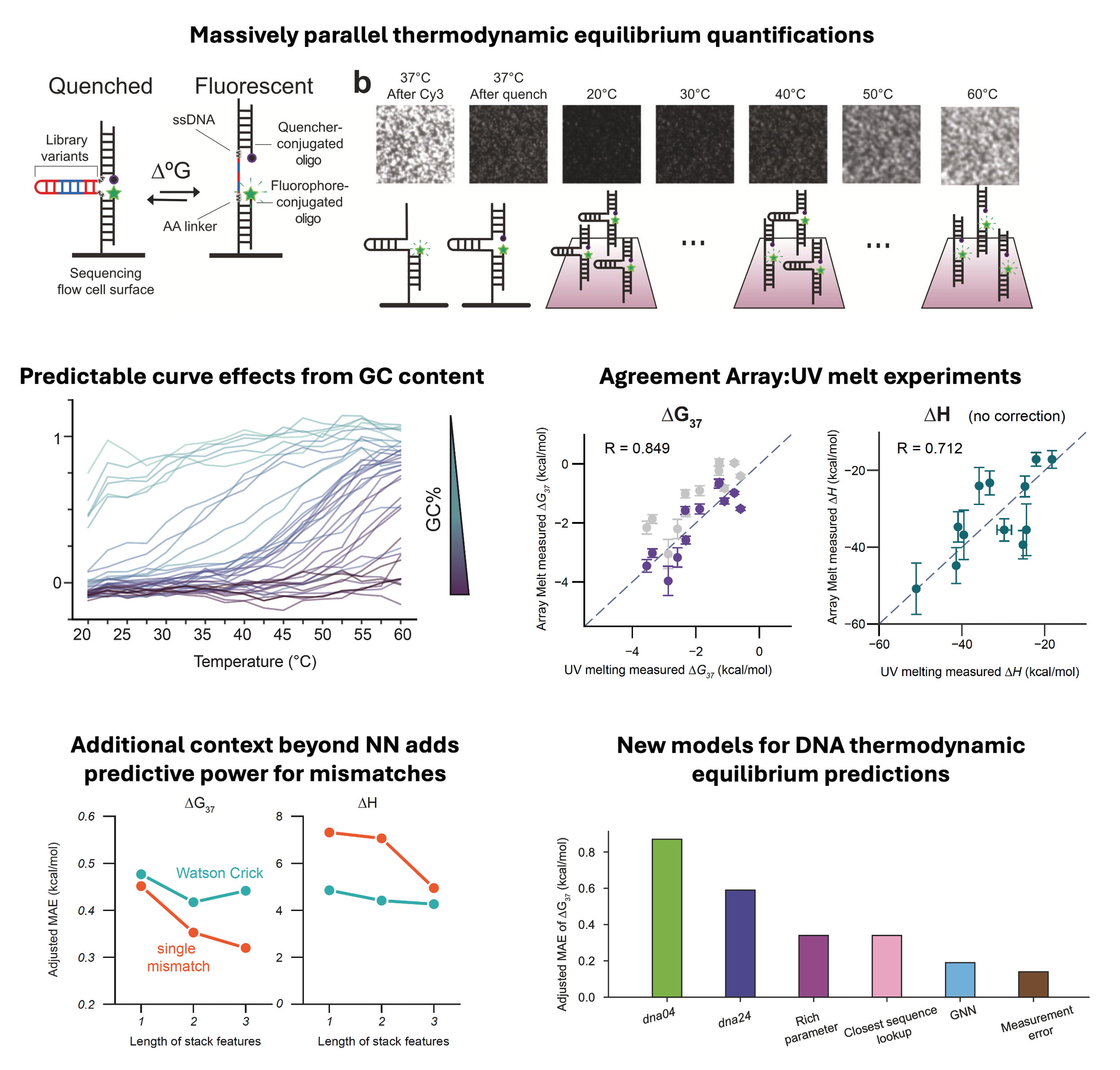
Yuxi Ke, Eesha Sharma, Hannah K Wayment-Steele, Winston R Becker, Anthony Ho, Emil Marklund, William J Greenleaf (2025) Nature Communications
High-throughput DNA melt measurements enable improved models of DNA folding thermodynamics
DNA folding thermodynamics are central to many biological processes and biotechnological applications involving base-pairing. Current methods for predicting stability from DNA sequence use nearest-neighbor models that struggle to accurately capture the diverse sequence dependence of secondary structural motifs beyond Watson-Crick base pairs, likely due to insufficient experimental data. In this work, we introduce a massively parallel method, Array Melt, that uses fluorescence-based quenching signals to measure the equilibrium stability of millions of DNA hairpins simultaneously on a repurposed Illumina sequencing flow cell. By leveraging this dataset of 27,732 sequences with two-state melting behaviors, we derive a NUPACK-compatible model (dna24), a rich parameter model that exhibits higher accuracy, and a graph neural network (GNN) model that identifies relevant interactions within DNA beyond nearest neighbors. All models show improved accuracy in predicting DNA folding thermodynamics, enabling more effective in silico design of qPCR primers, oligo hybridization probes, and DNA origami.
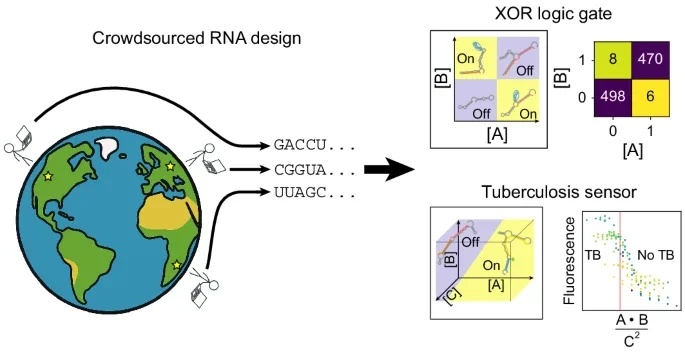
Christian A Choe, Johan OL Andreasson, Feriel Melaine, Wipapat Kladwang, Michelle J Wu, Fernando Portela, Roger Wellington-Oguri, John J Nicol, Hannah K Wayment-Steele, Michael Gotrik, Eterna Participants, Purvesh Khatri, William J Greenleaf, Rhiju Das (2025) Nature Chemistry
Compact RNA sensors for increasingly complex functions of multiple inputs
Designing single molecules that compute general functions of input molecular partners is a major unsolved challenge in molecular design. Here we demonstrate that high-throughput, iterative experimental testing of diverse RNA designs crowdsourced from the online game Eterna yields sensors of increasingly complex functions of input oligonucleotide concentrations. After designing single-input RNA sensors with activation ratios beyond our detection limits, we created logic gates, including challenging XOR and XNOR gates, and sensors that respond to the ratio of two inputs. Finally, we describe the OpenTB challenge, which elicited 85-nucleotide sensors that compute a score for diagnosing active tuberculosis based on the ratio of products of three gene segments. Building on OpenTB design strategies, we created an algorithm, Nucleologic, that produces similarly compact sensors for the three-gene score based on RNA and DNA. These results expand the possibilities for using compact, single-molecule sensors in a range of applications previously constrained by design complexity.
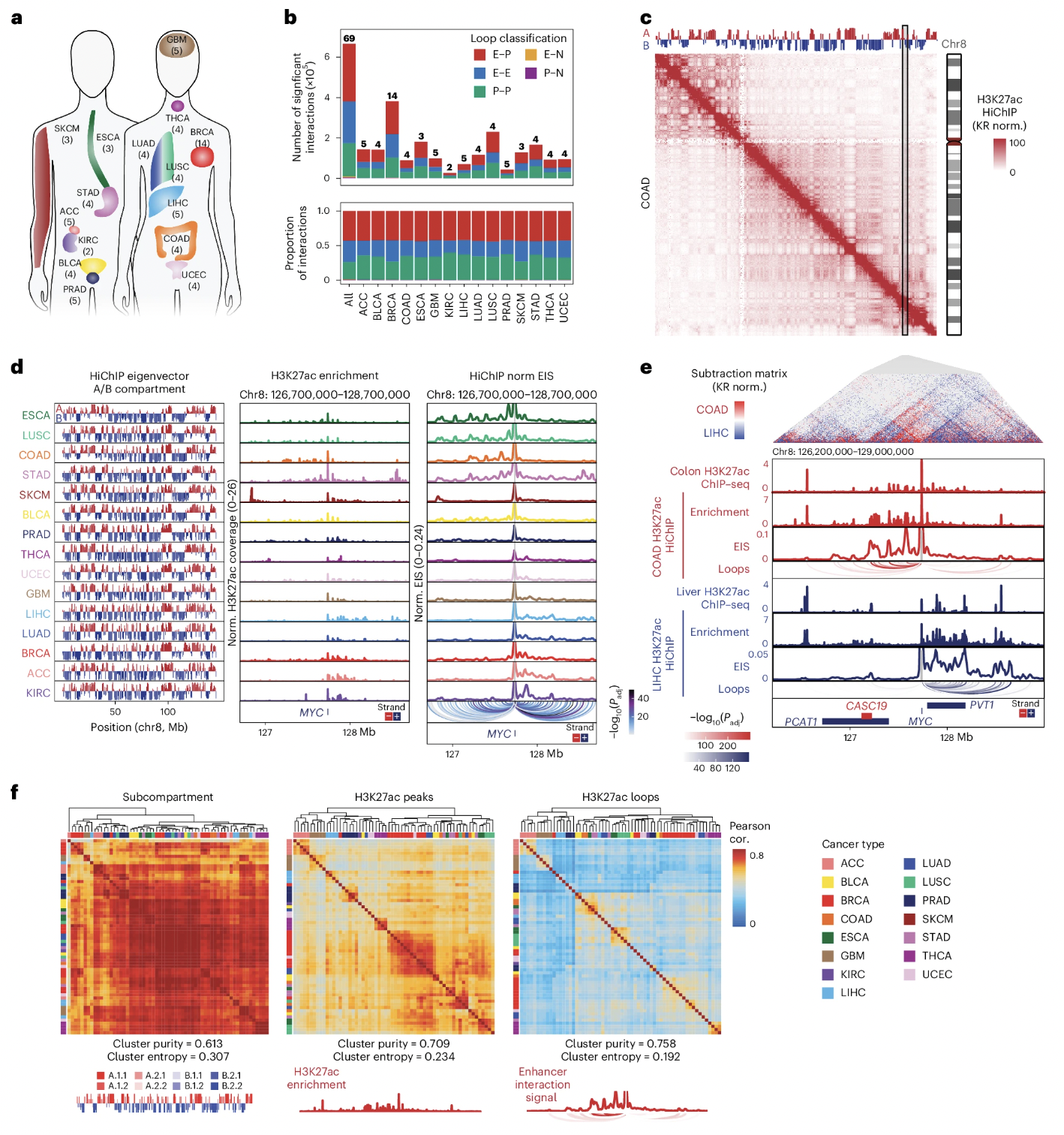
Kathryn E Yost, Yanding Zhao, King L Hung, Kaiyuan Zhu, Duo Xu, M Ryan Corces, Shadi Shams, Bryan H Louie, Shahab Sarmashghi, Laksshman Sundaram, Jens Luebeck, Stanley Clarke, Ashley S Doane, Jeffrey M Granja, Hani Choudhry, Marcin Imieliński, Andrew D Cherniack, Ekta Khurana, Vineet Bafna, Ina Felau, Jean C Zenklusen, Peter W Laird, Christina Curtis, William J Greenleaf, Howard Y Chang (2025) Nature Genetcs
Three-dimensional genome landscape of primary human cancers
Genome conformation underlies transcriptional regulation by distal enhancers, and genomic rearrangements in cancer can alter critical regulatory interactions. Here we profiled the three-dimensional genome architecture and enhancer connectome of 69 tumor samples spanning 15 primary human cancer types from The Cancer Genome Atlas. We discovered the following three archetypes of enhancer usage for over 100 oncogenes across human cancers: static, selective gain or dynamic rewiring. Integrative analyses revealed the enhancer landscape of noncancer cells in the tumor microenvironment for genes related to immune escape. Deep whole-genome sequencing and enhancer connectome mapping provided accurate detection and validation of diverse structural variants across cancer genomes and revealed distinct enhancer rewiring consequences from noncoding point mutations, genomic inversions, translocations and focal amplifications. Extrachromosomal DNA promoted more extensive enhancer rewiring among several types of focal amplification mechanisms. These results suggest a systematic approach to understanding genome topology in cancer etiology and therapy.
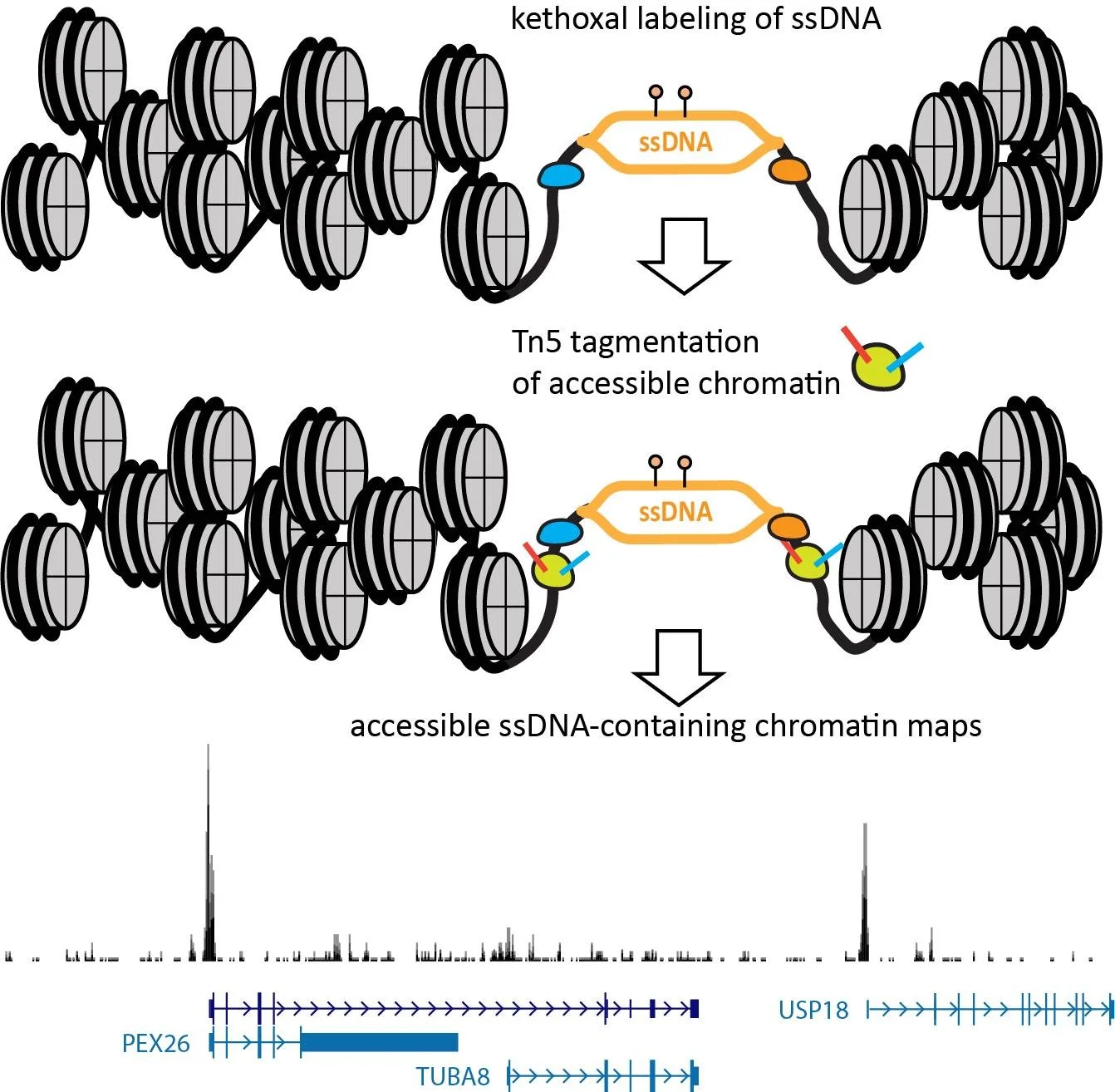
Georgi K Marinov, William J Greenleaf (2025) Bio-Protocol
Mapping the Simultaneously Accessible and ssDNA-Containing Genome With KAS-ATAC Sequencing
The KAS-ATAC assay provides a method to capture genomic DNA fragments that are simultaneously physically accessible and contain single-stranded DNA (ssDNA) bubbles. These are characteristic features of two of the key processes involved in regulating and expressing genes—on one hand, the activity of cis-regulatory elements (cREs), which are typically devoid of nucleosomes when active and occupied by transcription factors, and on the other, the association of RNA polymerases with DNA, which results in the presence of ssDNA structures. Here, we present a detailed protocol for carrying out KAS-ATAC as well as basic processing of KAS-ATAC datasets and discuss the key considerations for its successful application.
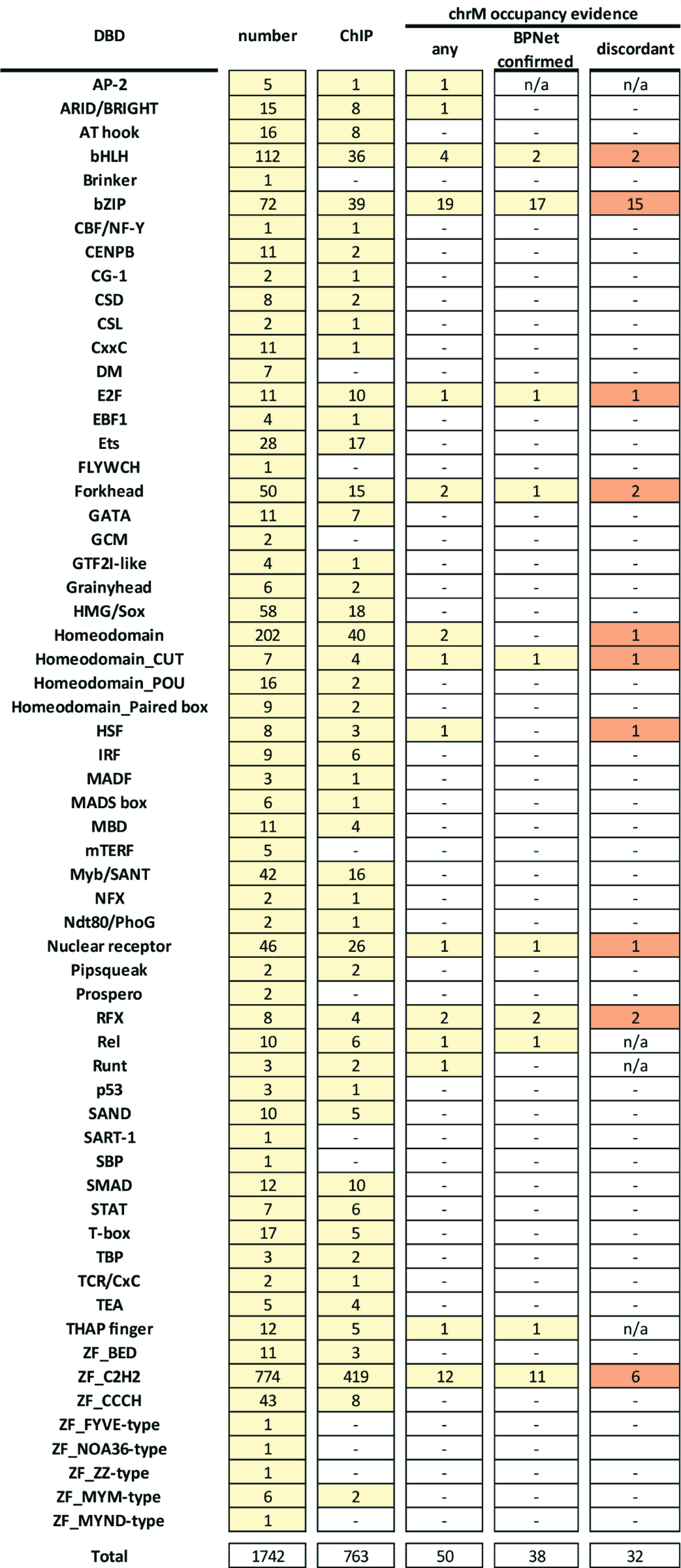
Georgi K Marinov, Vivekanandan Ramalingam, William J Greenleaf, Anshul Kundaje (2025) PloS One
An updated compendium and reevaluation of the evidence for nuclear transcription factor occupancy over the mitochondrial genome
In most eukaryotes, mitochondrial organelles contain their own genome, usually circular, which is the remnant of the genome of the ancestral bacterial endosymbiont that gave rise to modern mitochondria. Mitochondrial genomes are dramatically reduced in their gene content due to the process of endosymbiotic gene transfer to the nucleus; as a result most mitochondrial proteins are encoded in the nucleus and imported into mitochondria. This includes the components of the dedicated mitochondrial transcription and replication systems and regulatory factors, which are entirely distinct from the information processing systems in the nucleus. However, since the 1990s several nuclear transcription factors have been reported to act in mitochondria, and previously we identified 8 human and 3 mouse transcription factors (TFs) with strong localized enrichment over the mitochondrial genome using ChIP-seq (Chromatin Immunoprecipitation) datasets from the second phase of the ENCODE (Encyclopedia of DNA Elements) Project Consortium. Here, we analyze the greatly expanded in the intervening decade ENCODE compendium of TF ChIP-seq datasets (a total of 6,153 ChIP experiments for 942 proteins, of which 763 are sequence-specific TFs) combined with interpretative deep learning models of TF occupancy to create a comprehensive compendium of nuclear TFs that show evidence of association with the mitochondrial genome. We find some evidence for chrM occupancy for 50 nuclear TFs and two other proteins, with bZIP TFs emerging as most likely to be playing a role in mitochondria. However, we also observe that in cases where the same TF has been assayed with multiple antibodies and ChIP protocols, evidence for its chrM occupancy is not always reproducible. In the light of these findings, we discuss the evidential criteria for establishing chrM occupancy and reevaluate the overall compendium of putative mitochondrial-acting nuclear TFs.
More from 2025-26
Andreas R. Gschwind, Kristy S. Mualim, Alireza Karbalayghareh, Maya U. Sheth, Kushal K. Dey, Evelyn Jagoda, Ramil N. Nurtdinov, Wang Xi, Anthony S. Tan, James Galante, Hank Jones, X. Rosa Ma, David Yao, Dulguun Amgalan, Judhajeet Ray, Chad J. Munger, Joseph Nasser, Žiga Avsec, Benjamin T. James, Muhammad S. Shamim, Neva C. Durand, Suhas S. P. Rao, Ragini Mahajan, Benjamin R. Doughty, Kalina Andreeva, Jacob C. Ulirsch, Kaili Fan, Elizabeth M. Perez, Tri C. Nguyen, David R. Kelley, Hilary K. Finucane, Jill E. Moore, Zhiping Weng, Manolis Kellis, Michael C. Bassik, Berk Ustun, Alkes L. Price, Michael A. Beer, Roderic Guigó, John A. Stamatoyannopoulos, Erez Lieberman Aiden, William J. Greenleaf, Christina S. Leslie, Lars M. Steinmetz, Anshul Kundaje & Jesse M. Engreitz. An encyclopedia of human enhancer–gene regulatory interactions. Nature (2026). 10.1038/s41586-026-10781-4
Alma-Martina Cepika, Laura Amaya, Colin Waichler, Mansi Narula, Michelle Mantilla, Benjamin C Thomas, Pauline P Chen, Robert A Freeborn, Mara Pavel-Dinu, Jason Nideffer, Matthew Porteus, Rosa Bacchetta, Fabian Müller, William J Greenleaf, Howard Y Chang, Maria Grazia Roncarolo. Epigenetic landscape, key transcriptional regulators, and in vivo identification of human Tr1 cells. Science Advances (2026). doi: 10.1126/sciadv.aec6358
Debra Van Egeren, Ryan O Schenck, Aziz Khan, Aaron M Horning, Shanlan Mo, Clemens L Weiß, Edward D Esplin, Winston R Becker, Si Wu, Casey Hanson, Nasim Barapour, Lihua Jiang, Kévin Contrepois, Hayan Lee, Stephanie A Nevins, Tuhin K Guha, Hao Zhang, Zhen He, Zhicheng Ma, Emma Monte, Thomas V Karathanos, Rozelle Laquindanum, Meredith A Mills, Hassan Chaib, Roxanne Chiu, Ruiqi Jian, Joanne Chan, Mathew Ellenberger, Bahareh Bahmani, Basil Michael, Annika K Weimer, D Glen Esplin, Samuel Lancaster, Jeanne Shen, Uri Ladabaum, Teri A Longacre, Anshul Kundaje, William J Greenleaf, Zheng Hu, James M Ford, Michael P Snyder, Christina Curtis. Polyclonal origins of human premalignant colorectal lesions. Nature (2025). doi: 10.1038/s41586-025-09930-y.
William Thistlethwaite, Sindhu Vangeti, Wan-Sze Cheng, Pankaj Agarwal, Antonio Cappuccio, Wenliang Wang, Bei Wei, Rachel Myers, Aliza B Rubenstein, Daniel Chawla, Manoj Hariharan, Micah T McClain, Thomas W Burke, Steven H Kleinstein, Joseph R Ecker, Christopher W Woods, William J Greenleaf, Xi Chen, Irene Ramos, Elena Zaslavsky, Thomas G Evans, Olga G Troyanskaya, Stuart C Sealfon. Innate immune molecular landscape following controlled human influenza virus infection. Cell Reports (2025). doi: 10.1016/j.celrep.2025.116312.
Yuning J Tang, Haiqing Xu, Nicholas W Hughes, Paloma Ruiz, Samuel H Kim, Emily G Shuldiner, Steven S Lopez, Jess D Hebert, Saswati Karmakar, Laura Andrejka, Deniz Nesli Dolcen, Gabor Boross, Pauline Chu, Christian A Kunder, Colin Detrick, Sarah E Pierce, Emily L Ashkin, William J Greenleaf, Anne K Voss, Tim Thomas, Matt van de Rijn, Dmitri A Petrov, Monte M Winslow. Functional mapping of epigenomic regulators uncovers coordinated tumor suppression by the HBO1 and MLL1 complexes. Cancer Discovery (2025). doi: 10.1158/2159-8290.CD-24-1565.
Noah Gamble, Jason A Caldwell, Joshua McKeever, Caroline Kaiser, Alexandra Bradu, Peyton J Dooley, Sandy Klemm, William J Greenleaf, Narutoshi Hibino, Aaron R Dinner, Andrew S Koh. Thymic epithelial cells amplify epigenetic noise to promote immune tolerance. Nature (2025). doi: 10.1038/s41586-025-09424-x
Caleb A Lareau, Patrick Maschmeyer, Yajie Yin, Jacob C Gutierrez, Ryan S Dhindsa, Anne-Sophie Gribling-Burrer, Sebastian Zielinski, Yu-Hsin Hsieh, Lena Nitsch, Veronika Dimitrova, Benan Nalbant, Frank A Buquicchio, Tsion Abay, Robert R Stickels, Jacob C Ulirsch, Patrick Yan, Fangyi Wang, Zhuang Miao, Katalin Sandor, Bence Daniel, Vincent Liu, Paul L Mendez, Petra Knaus, Manpreet Meyer, William J Greenleaf, Anshul Kundaje, Redmond P Smyth, Mathias Munschauer, Leif S Ludwig, Ansuman T Satpathy. Cell type-specific purifying selection of synonymous mitochondrial DNA variation. PNAS (2025). doi: 10.1073/pnas.2505704122.
Mazhar Adli, Laralynne Przybyla, Tony Burdett, Paul W Burridge, Pilar Cacheiro, Howard Y Chang, Jesse M Engreitz, Luke A Gilbert, William J Greenleaf, Li Hsu, Danwei Huangfu, Ling-Hong Hung, Anshul Kundaje, Sheng Li, Helen Parkinson, Xiaojie Qiu, Paul Robson, Stephan C Schürer, Ali Shojaie, William C Skarnes, Damian Smedley, Lorenz Studer, Wei Sun, Dušica Vidović, Thomas Vierbuchen, Brian S White, Ka Yee Yeung, Feng Yue, Ting Zhou. MorPhiC Consortium: towards functional characterization of all human genes. Nature (2025). doi: 10.1038/s41586-024-08243-w.
2024 and beyond

Benjamin R. Doughty, Michaela M. Hinks, Julia M. Schaepe, Georgi K. Marinov, Abby R. Thurm, Carolina Rios-Martinez, Benjamin E. Parks, Yingxuan Tan, Emil Marklund, Danilo Dubocanin, Lacramioara Bintu, William J. Greenleaf (2024) Nature
Single-molecule states link transcription factor binding to gene expression
The binding of multiple transcription factors (TFs) to genomic enhancers drives gene expression in mammalian cells. However, the molecular details that link enhancer sequence to TF binding, promoter state and transcription levels remain unclear. Here we applied single-molecule footprinting to measure the simultaneous occupancy of TFs, nucleosomes and other regulatory proteins on engineered enhancer–promoter constructs with variable numbers of TF binding sites for both a synthetic TF and an endogenous TF involved in the type I interferon response. Although TF binding events on nucleosome-free DNA are independent, activation domains recruit cofactors that destabilize nucleosomes, driving observed TF binding cooperativity. Average TF occupancy linearly determines promoter activity, and we decompose TF strength into separable binding and activation terms. Finally, we develop thermodynamic and kinetic models that quantitatively predict both the enhancer binding microstates and gene expression dynamics. This work provides a template for the quantitative dissection of distinct contributors to gene expression, including TF activation domains, concentration, binding affinity, binding site configuration and recruitment of chromatin regulators.
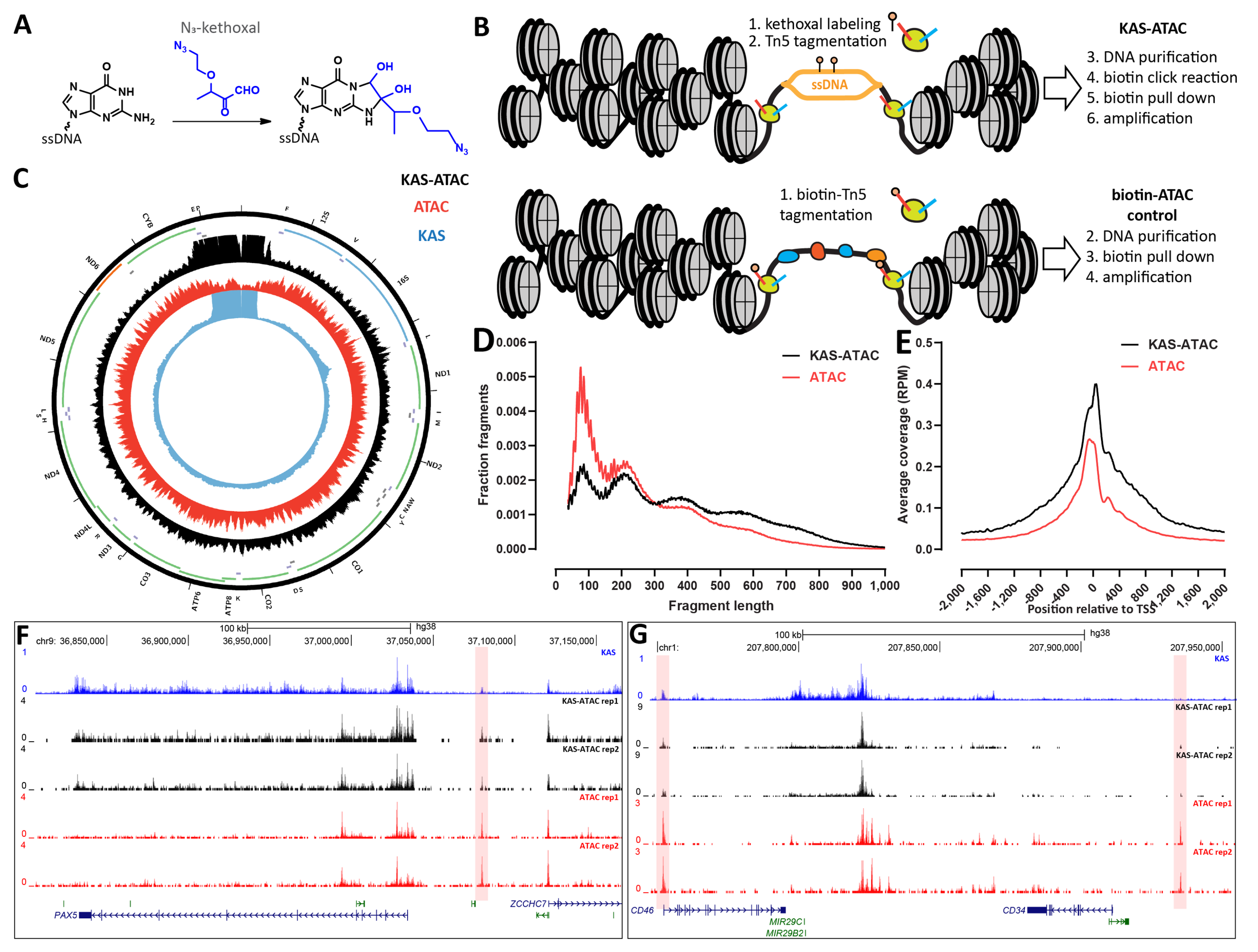
Samuel H. Kim, Georgi K. Marinov, William J. Greenleaf (2024) Genome Research
KAS-ATAC reveals the genome-wide single-stranded accessible chromatin landscape of the human genome
Gene regulation in most eukaryotes involves two fundamental processes – alterations in genome packaging by nucleosomes, with active cis-regulatory elements (CREs) generally characterized by open-chromatin configuration, and transcriptional activation. Mapping these physical properties and biochemical activities – through profiling chromatin accessibility and active transcription – are key tools for understanding the logic and mechanisms of transcription and its regulation. However, the relationship between these two states has not been accessible to simultaneous measurement. To this end, we developed KAS-ATAC, a combination of the KAS-seq (Kethoxal- Assisted SsDNA sequencing and ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) methods for mapping single-stranded DNA (and thus active transcription) and chromatin accessibility, respectively, enabling the genome-wide identification of DNA fragments that are simultaneously accessible and contain ssDNA. We use KAS-ATAC to evaluate levels of ac- tive transcription over different CRE classes, to estimate absolute levels of transcribed accessible DNA over CREs, to map nucleosomal configurations associated with RNA polymerase activities, and to assess transcription factor association with transcribed DNA through transcription factor binding site (TFBS) footprinting. We observe lower levels of transcription over distal enhancers compared to promoters and distinct nucleosomal configurations around transcription initiation sites associated with active transcription. We find that most TFs associate equally with transcribed and non-transcribed DNA but a few factors specifically do not exhibit footprints over ssDNA-containing fragments. We anticipate KAS-ATAC to continue to derive useful in- sights into chromatin organization and transcriptional regulation in other contexts in the future.
Georgi K. Marinov, Benjamin Doughty, Anshul Kundaje, William J. Greenleaf (2024) Genome Research
The chromatin landscape of the histone-possessing Bacteriovorax bacteria
Histone proteins have traditionally been thought to be restricted to eukaryotes and most archaea, with eukaryotic nucleosomal histones deriving from their archaeal ancestors. In contrast, bacteria lack histones as a rule. However, histone proteins have recently been identified in a few bacterial clades, most notably the phylum Bdellovibrionota, and these histones have been proposed to exhibit a range of divergent features compared to histones in archaea and eukaryotes. However, no functional genomic studies of the properties of Bdellovibrionota chromatin have been carried out. In this work, we map the landscape of chromatin accessibility, active transcription and three-dimensional genome organization in a member of Bdellovibrionota (a Bacteriovorax strain). We find that, similar to what is observed in some archaea and in eukaryotes with compact genomes such as yeast, Bacteriovorax chromatin is characterized by preferential accessibility around promoter regions. Similar to eukaryotes, chromatin accessibility in Bacteriovorax positively correlates with gene expression. Mapping active transcription through single-strand DNA (ssDNA) profiling revealed that unlike in yeast, but similar to the state of mammalian and fly promoters, Bacteriovorax promoters exhibit very strong polymerase pausing. Finally, similar to that of other bacteria without histones, the Bacteriovorax genome exists in a three-dimensional (3D) configuration organized by the parABS system along the axis defined by replication origin and termination regions. These results provide a foundation for understanding the chromatin biology of the unique Bdellovibrionota bacteria and the functional diversity in chromatin organization across the tree of life.
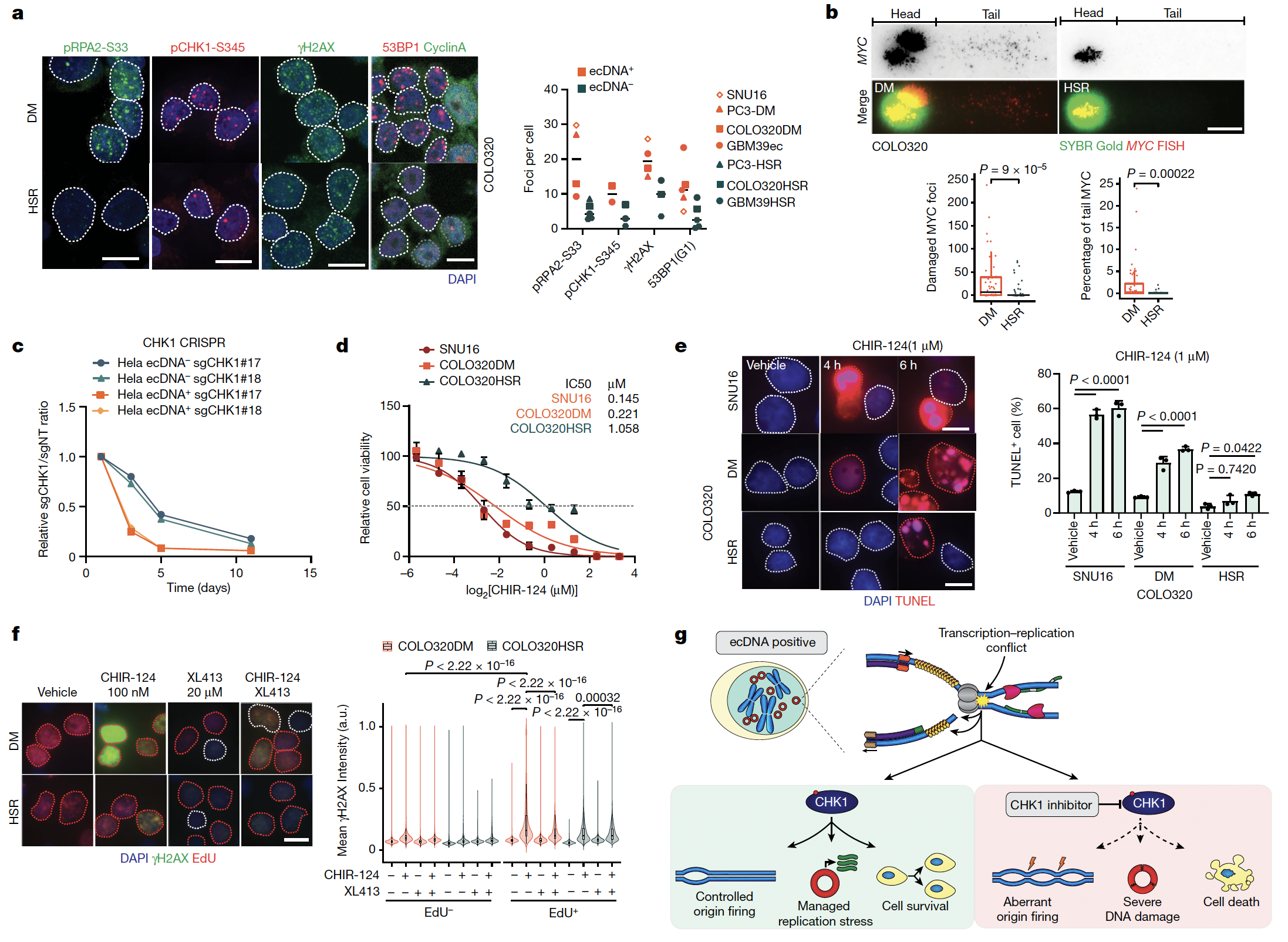
Jun Tang, Natasha E Weiser, Guiping Wang, Sudhir Chowdhry, Ellis J Curtis, Yanding Zhao, Ivy Tsz-Lo Wong, Georgi K Marinov, Rui Li, Philip Hanoian, Edison Tse, Salvador Garcia Mojica, Ryan Hansen, Joshua Plum, Auzon Steffy, Snezana Milutinovic, S Todd Meyer, Jens Luebeck, Yanbo Wang, Shu Zhang, Nicolas Altemose, Christina Curtis, William J Greenleaf, Vineet Bafna, Stephen J Benkovic, Anthony B Pinkerton, Shailaja Kasibhatla, Christian A Hassig, Paul S Mischel, Howard Y Chang (2024) Nature
Enhancing transcription–replication conflict targets ecDNA-positive cancers
Extrachromosomal DNA (ecDNA) presents a major challenge for cancer patients. ecDNA renders tumours treatment resistant by facilitating massive oncogene transcription and rapid genome evolution, contributing to poor patient survival. At present, there are no ecDNA-specific treatments. Here we show that enhancing transcription–replication conflict enables targeted elimination of ecDNA-containing cancers. Stepwise analyses of ecDNA transcription reveal pervasive RNA transcription and associated single-stranded DNA, leading to excessive transcription–replication conflicts and replication stress compared with chromosomal loci. Nucleotide incorporation on ecDNA is markedly slower, and replication stress is significantly higher in ecDNA-containing tumours regardless of cancer type or oncogene cargo. pRPA2-S33, a mediator of DNA damage repair that binds single-stranded DNA, shows elevated localization on ecDNA in a transcription-dependent manner, along with increased DNA double strand breaks, and activation of the S-phase checkpoint kinase, CHK1. Genetic or pharmacological CHK1 inhibition causes extensive and preferential tumour cell death in ecDNA-containing tumours. We advance a highly selective, potent and bioavailable oral CHK1 inhibitor, BBI-2779, that preferentially kills ecDNA-containing tumour cells. In a gastric cancer model containing FGFR2 amplified on ecDNA, BBI-2779 suppresses tumour growth and prevents ecDNA-mediated acquired resistance to the pan-FGFR inhibitor infigratinib, resulting in potent and sustained tumour regression in mice. Transcription–replication conflict emerges as a target for ecDNA-directed therapy, exploiting a synthetic lethality of excess to treat cancer.
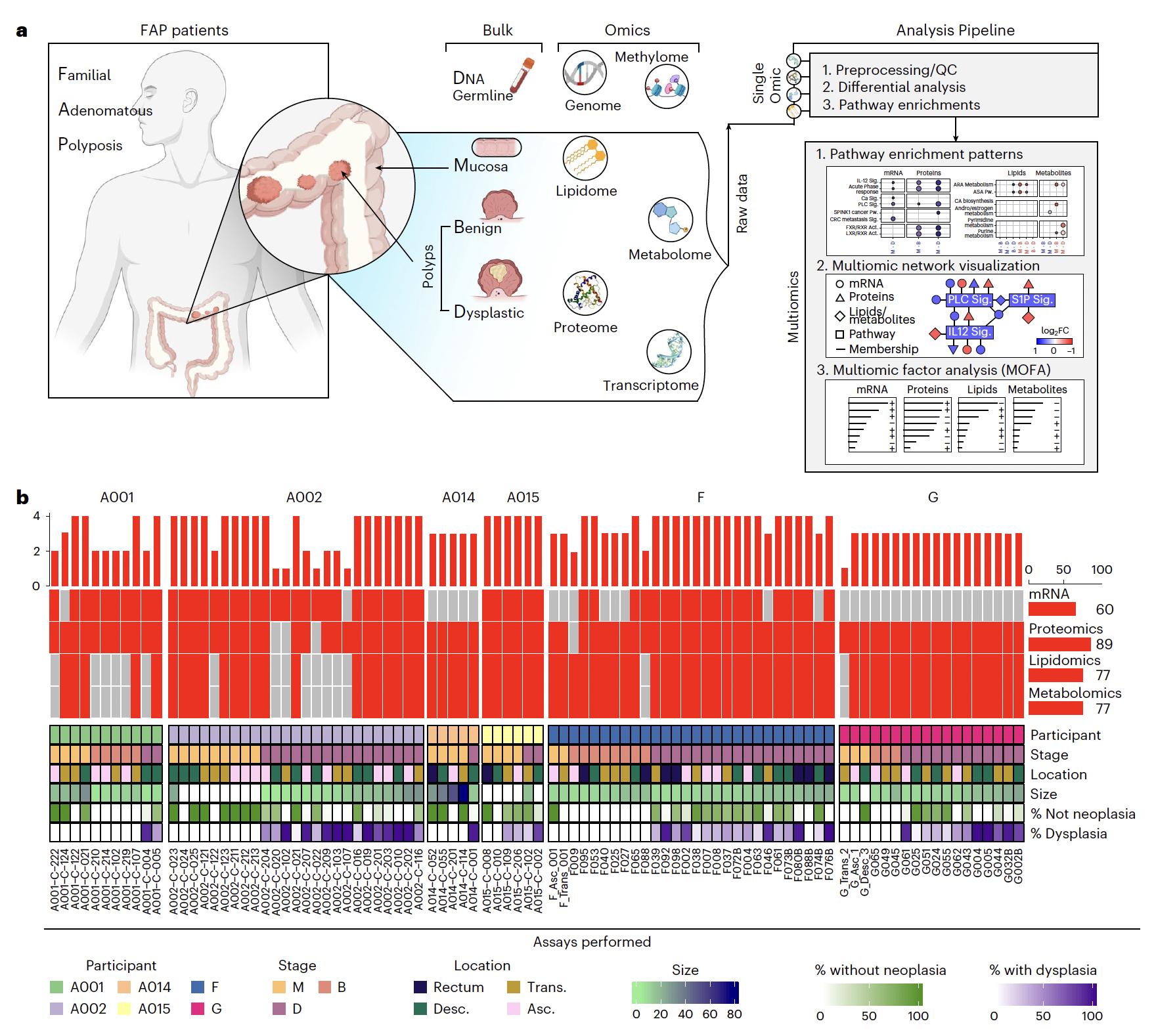
Edward D Esplin, Casey Hanson, Si Wu, Aaron M Horning, Nasim Barapour, Stephanie A Nevins, Lihua Jiang, Kévin Contrepois, Hayan Lee, Tuhin K Guha, Zheng Hu, Rozelle Laquindanum, Meredith A Mills, Hassan Chaib, Roxanne Chiu, Ruiqi Jian, Joanne Chan, Mathew Ellenberger, Winston R Becker, Bahareh Bahmani, Aziz Khan, Basil Michael, Annika K Weimer, D Glen Esplin, Jeanne Shen, Samuel Lancaster, Emma Monte, Thomas V Karathanos, Uri Ladabaum, Teri A Longacre, Anshul Kundaje, Christina Curtis, William J Greenleaf, James M Ford, Michael P Snyder (2024) Nature Cancer
Multiomic analysis of familial adenomatous polyposis reveals molecular pathways associated with early tumorigenesis
Familial adenomatous polyposis (FAP) is a genetic disease causing hundreds of premalignant polyps in affected persons and is an ideal model to study transitions of early precancer states to colorectal cancer (CRC). We performed deep multiomic profiling of 93 samples, including normal mucosa, benign polyps and dysplastic polyps, from six persons with FAP. Transcriptomic, proteomic, metabolomic and lipidomic analyses revealed a dynamic choreography of thousands of molecular and cellular events that occur during precancerous transitions toward cancer formation. These involve processes such as cell proliferation, immune response, metabolic alterations (including amino acids and lipids), hormones and extracellular matrix proteins. Interestingly, activation of the arachidonic acid pathway was found to occur early in hyperplasia; this pathway is targeted by aspirin and other nonsteroidal anti-inflammatory drugs, a preventative treatment under investigation in persons with FAP. Overall, our results reveal key genomic, cellular and molecular events during the earliest steps in CRC formation and potential mechanisms of pharmaceutical prophylaxis.
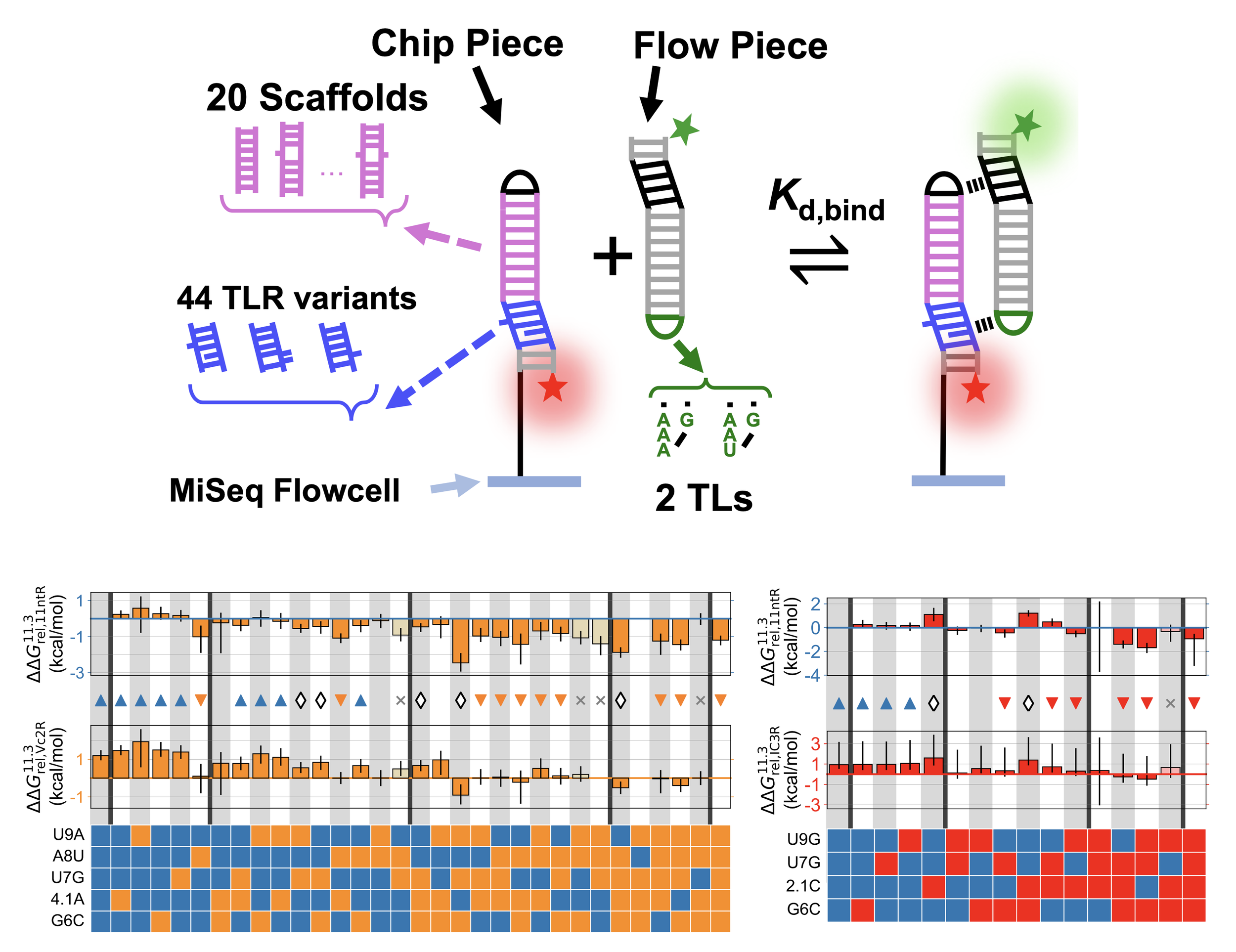
John H. Shin, Lena M. Cuevas, Rohit Roy, Steve L. Bonilla, Hashim Al-Hashimi, William J Greenleaf, Daniel Herschlag (2024) RNA
Exploring the energetic and conformational properties of the sequence space connecting naturally occurring RNA tetraloop receptor motifs
Folded RNAs contain tertiary contact motifs whose structures and energetics are conserved across different RNAs. The transferable properties of RNA motifs simplify the RNA folding problem, but measuring energetic and conformational properties of many motifs remains a challenge. Here, we use a high-throughput thermodynamic approach to investigate how sequence changes alter the binding properties of naturally-occurring motifs, the GAAA tetraloop • tetraloop receptor (TLR) interactions. We measured the binding energies and conformational preferences of TLR sequences that span mutational pathways from the canonical 11ntR to two other natural TLRs, the IC3R and Vc2R. While the IC3R and Vc2R share highly similar energetic and conformational properties, the landscapes that map the sequence changes for their conversion from the 11ntR to changes in these properties differ dramatically. Differences in the energetic landscapes stem from the mutations needed to convert the 11ntR to the IC3R and Vc2R rather than a difference in the intrinsic energetic architectures of these TLRs. The conformational landscapes feature several non-native TLR variants with conformational preferences that differ from both the initial and final TLRs; these species represent potential branching points along the multidimensional sequence space to sequences with greater fitness in other RNA contexts with alternative conformational preferences. Our high-throughput, quantitative approach reveals the complex nature of sequence-fitness landscapes and leads to models for their molecular origins. Systematic and quantitative molecular approaches provide critical insights into understanding the evolution of natural RNAs as they traverse complex landscapes in response to selective pressures.
Yizhou Zhu, Hayan Lee, Shannon White, Annika K Weimer, Emma Monte, Aaron Horning, Stephanie A Nevins, Edward D Esplin, Kristina Paul, Gat Krieger, Zohar Shipony, Roxanne Chiu, Rozelle Laquindanum, Thomas V Karathanos, Melissa WY Chua, Meredith Mills, Uri Ladabaum, Teri Longacre, Jeanne Shen, Ariel Jaimovich, Doron Lipson, Anshul Kundaje, William J Greenleaf, Christina Curtis, James M Ford, Michael P Snyder (2024) Nature Cancer
Global loss of promoter–enhancer connectivity and rebalancing of gene expression during early colorectal cancer carcinogenesis
Although three-dimensional (3D) genome architecture is crucial for gene regulation, its role in disease remains elusive. We traced the evolution and malignant transformation of colorectal cancer (CRC) by generating high-resolution chromatin conformation maps of 33 colon samples spanning different stages of early neoplastic growth in persons with familial adenomatous polyposis (FAP). Our analysis revealed a substantial progressive loss of genome-wide cis-regulatory connectivity at early malignancy stages, correlating with nonlinear gene regulation effects. Genes with high promoter–enhancer (P–E) connectivity in unaffected mucosa were not linked to elevated baseline expression but tended to be upregulated in advanced stages. Inhibiting highly connected promoters preferentially represses gene expression in CRC cells compared to normal colonic epithelial cells. Our results suggest a two-phase model whereby neoplastic transformation reduces P–E connectivity from a redundant state to a rate-limiting one for transcriptional levels, highlighting the intricate interplay between 3D genome architecture and gene regulation during early CRC progression.
Noah Gamble, Alexandra Bradu, Jason A. Caldwell, Joshua McKeever, Olubusayo Bolonduro, Ebru Ermis, Caroline Kaiser, YeEun Kim, Benjamin Parks, Sandy Klemm, William J. Greenleaf, Gerald R. Crabtree, Andrew S. Koh (2024) Nature Immunology
PU.1 and BCL11B sequentially cooperate with RUNX1 to anchor mSWI/SNF to poise the T cell effector landscape
Adaptive immunity relies on specialized effector functions elicited by lymphocytes, yet how antigen recognition activates appropriate effector responses through nonspecific signaling intermediates is unclear. Here we examined the role of chromatin priming in specifying the functional outputs of effector T cells and found that most of the cis-regulatory landscape active in effector T cells was poised early in development before the expression of the T cell antigen receptor. We identified two principal mechanisms underpinning this poised landscape: the recruitment of the nucleosome remodeler mammalian SWItch/Sucrose Non-Fermentable (mSWI/SNF) by the transcription factors RUNX1 and PU.1 to establish chromatin accessibility at T effector loci; and a ‘relay’ whereby the transcription factor BCL11B succeeded PU.1 to maintain occupancy of the chromatin remodeling complex mSWI/SNF together with RUNX1, after PU.1 silencing during lineage commitment. These mechanisms define modes by which T cells acquire the potential to elicit specialized effector functions early in their ontogeny and underscore the importance of integrating extrinsic cues to the developmentally specified intrinsic program.
Georgi K. Marinov, Xinyi Chen, Matthew P. Swaffer, Tingting Xiang, Arthur R. Grossman, William J. Greenleaf (2024) Genome Biology
Genome-wide distribution of 5-hydroxymethyluracil and chromatin accessibility in the Breviolum minutum genome
In dinoflagellates, a unique and extremely divergent genomic and nuclear organization has evolved. The highly unusual features of dinoflagellate nuclei and genomes include permanently condensed liquid crystalline chromosomes, primarily packaged by proteins other than histones, genes organized in very long unidirectional gene arrays, a general absence of transcriptional regulation, high abundance of the otherwise very rare DNA modification 5-hydroxymethyluracil (5-hmU), and many others. While most of these fascinating properties are originally identified in the 1970s and 1980s, they have not yet been investigated using modern genomic tools. In this work, we address some of the outstanding questions regarding dinoflagellate genome organization by mapping the genome-wide distribution of 5-hmU (using both immunoprecipitation-based and basepair-resolution chemical mapping approaches) and of chromatin accessibility in the genome of the Symbiodiniaceae dinoflagellate Breviolum minutum. We find that the 5-hmU modification is preferentially enriched over certain classes of repetitive elements, often coincides with the boundaries between gene arrays, and is generally correlated with decreased chromatin accessibility, the latter otherwise being largely uniform along the genome. We discuss the potential roles of 5-hmU in the functional organization of dinoflagellate genomes and its relationship to the transcriptional landscape of gene arrays. Our results provide the first window into the 5-hmU and chromatin accessibility landscapes in dinoflagellates.
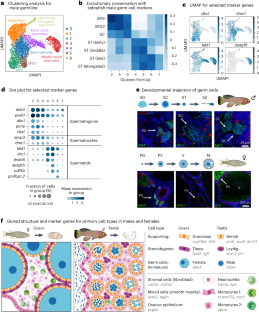
Eitan Moses, Tehila Atlan, Xue Sun, Roman Franěk, Atif Siddiqui, Georgi K. Marinov, Sagiv Shifman, David M. Zucker, Adi Oron-Gottesman, William J. Greenleaf, Ehud Cohen, Oren Ram, Itamar Harel (2024) Nature Aging
The killifish germline regulates longevity and somatic repair in a sex-specific manner
Classical evolutionary theories propose tradeoffs among reproduction, damage repair and lifespan. However, the specific role of the germline in shaping vertebrate aging remains largely unknown. In this study, we used the turquoise killifish (Nothobranchius furzeri) to genetically arrest germline development at discrete stages and examine how different modes of infertility impact life history. We first constructed a comprehensive single-cell gonadal atlas, providing cell-type-specific markers for downstream phenotypic analysis. We show here that germline depletion—but not arresting germline differentiation—enhances damage repair in female killifish. Conversely, germline-depleted males instead showed an extension in lifespan and rejuvenated metabolic functions. Through further transcriptomic analysis, we highlight enrichment of pro-longevity pathways and genes in germline-depleted male killifish and demonstrate functional conservation of how these factors may regulate longevity in germline-depleted Caenorhabditis elegans. Our results, therefore, demonstrate that different germline manipulation paradigms can yield pronounced sexually dimorphic phenotypes, implying alternative responses to classical evolutionary tradeoffs.
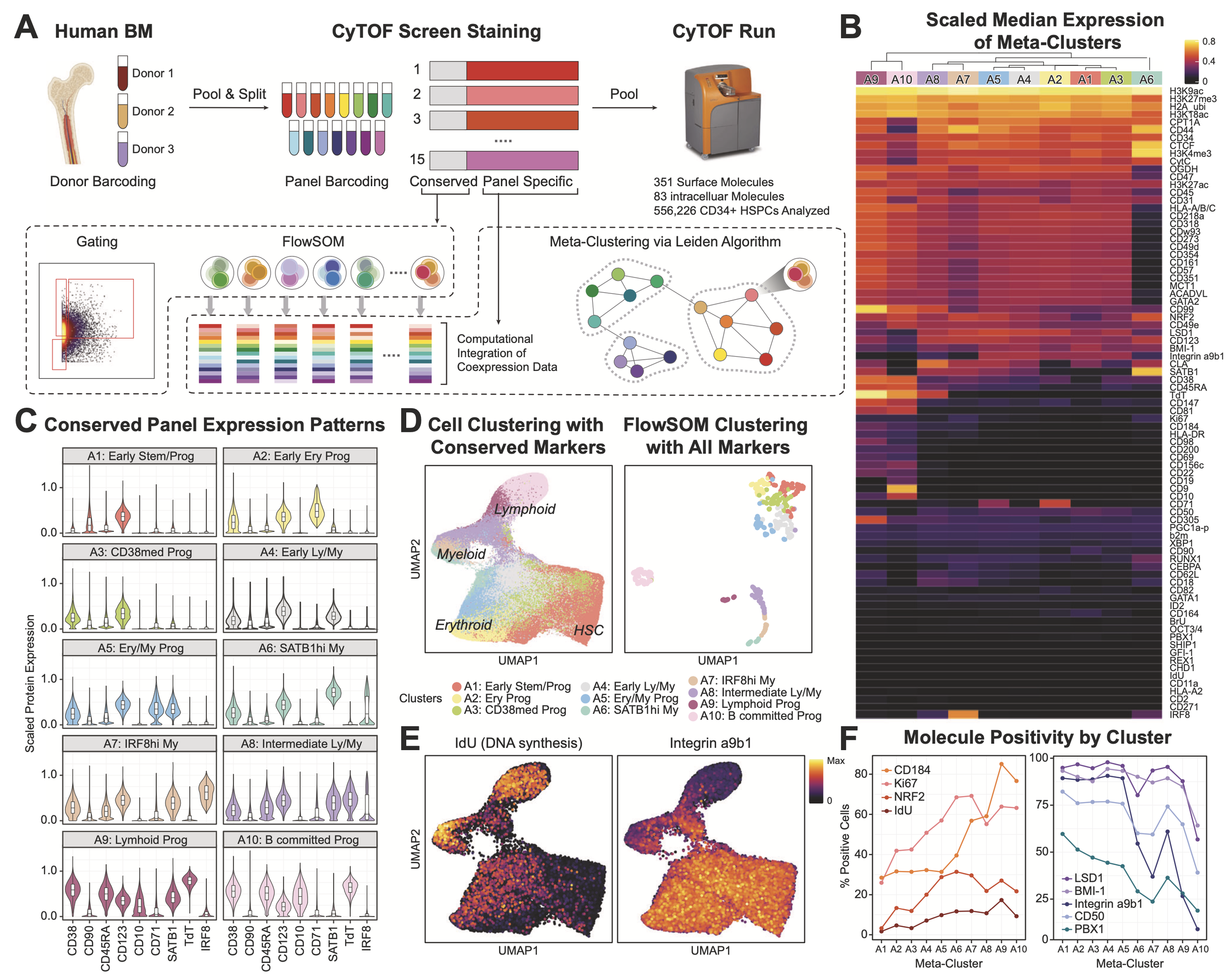
YeEun Kim, Ariel A. Calderon, Patricia Favaro, David R. Glass, Albert G. Tsai, Daniel Ho, Luciene Borges, William J. Greenleaf, Sean C. Bendall (2024) Nature Communications
Terminal deoxynucleotidyl transferase and CD84 identify human multi-potent lymphoid progenitors
Lymphoid specification in human hematopoietic progenitors is not fully understood. To better associate lymphoid identity with protein-level cell features, we conduct a highly multiplexed single-cell proteomic screen on human bone marrow progenitors. This screen identifies terminal deoxynucleotidyl transferase (TdT), a specialized DNA polymerase intrinsic to VDJ recombination, broadly expressed within CD34+ progenitors prior to B/T cell emergence. While these TdT+ cells coincide with granulocyte-monocyte progenitor (GMP) immunophenotype, their accessible chromatin regions show enrichment for lymphoid-associated transcription factor (TF) motifs. TdT expression on GMPs is inversely related to the SLAM family member CD84. Prospective isolation of CD84lo GMPs demonstrates robust lymphoid potentials ex vivo, while still retaining significant myeloid differentiation capacity, akin to LMPPs. This multi-omic study identifies human bone marrow lymphoid-primed progenitors, further defining the lympho-myeloid axis in human hematopoiesis.
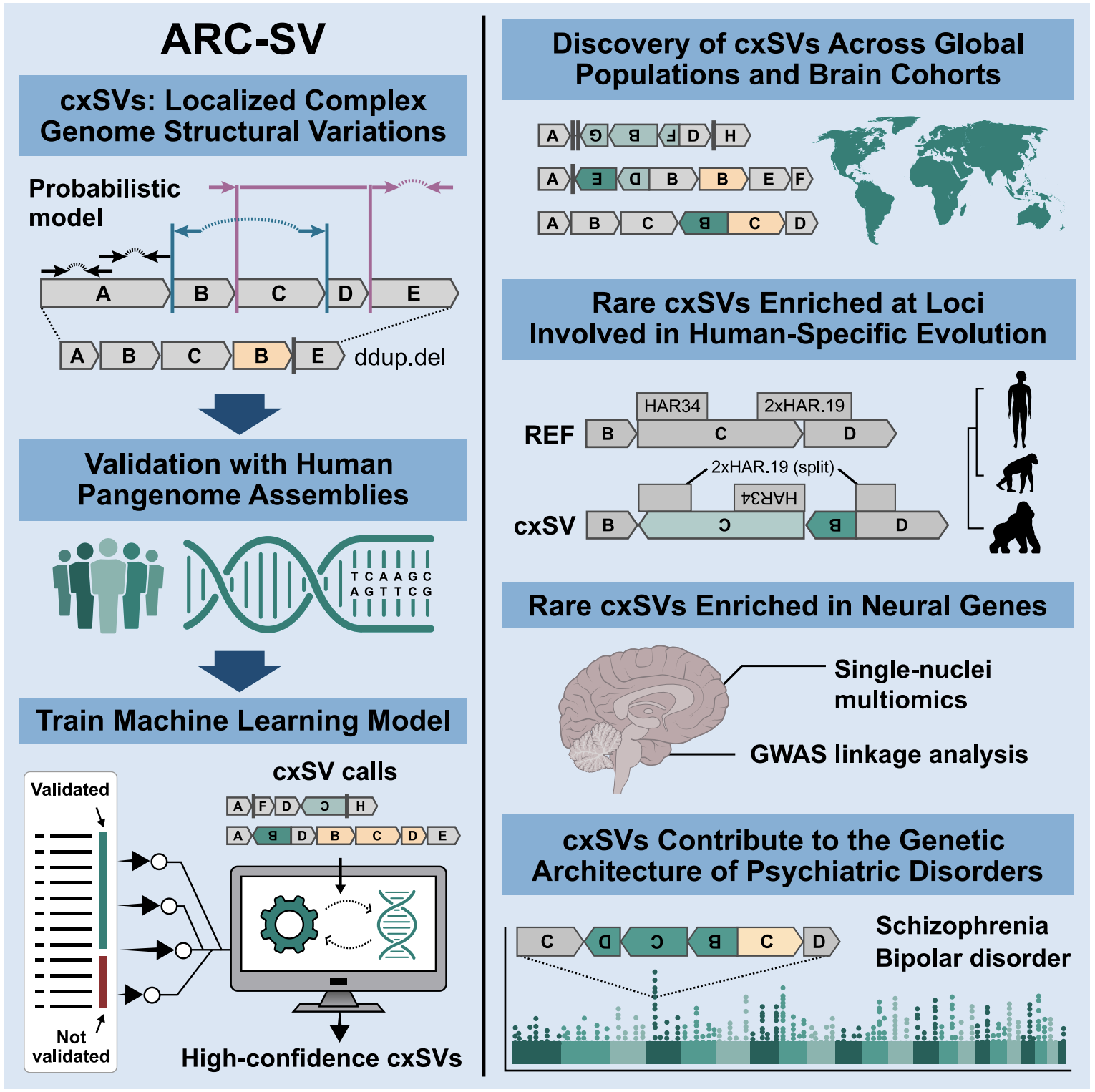
Bo Zhou, Joseph G Arthur, Hanmin Guo, Taeyoung Kim, Yiling Huang, Reenal Pattni, Tao Wang, Soumya Kundu, Jay XJ Luo, HoJoon Lee, Daniel C Nachun, Carolin Purmann, Emma M Monte, Annika K Weimer, Ping-Ping Qu, Minyi Shi, Lixia Jiang, Xinqiong Yang, John F Fullard, Jaroslav Bendl, Kiran Girdhar, Minsu Kim, Xi Chen, William J Greenleaf, Laramie Duncan, Hanlee P Ji, Xiang Zhu, Giltae Song, Stephen B Montgomery, Dean Palejev, Heinrich Zu Dohna, Panos Roussos, Anshul Kundaje, Joachim F Hallmayer, Michael P Snyder, Wing H Wong, Alexander E Urban (2024) Cell
Detection and analysis of complex structural variation in human genomes across populations and in brains of donors with psychiatric disorders
Complex structural variations (cxSVs) are often overlooked in genome analyses due to detection challenges. We developed ARC-SV, a probabilistic and machine-learning-based method that enables accurate detection and reconstruction of cxSVs from standard datasets. By applying ARC-SV across 4,262 genomes representing all continental populations, we identified cxSVs as a significant source of natural human genetic variation. Rare cxSVs have a propensity to occur in neural genes and loci that underwent rapid human-specific evolution, including those regulating corticogenesis. By performing single-nucleus multiomics in postmortem brains, we discovered cxSVs associated with differential gene expression and chromatin accessibility across various brain regions and cell types. Additionally, cxSVs detected in brains of psychiatric cases are enriched for linkage with psychiatric GWAS risk alleles detected in the same brains. Furthermore, our analysis revealed significantly decreased brain-region- and cell-type-specific expression of cxSV genes, specifically for psychiatric cases, implicating cxSVs in the molecular etiology of major neuropsychiatric disorders.
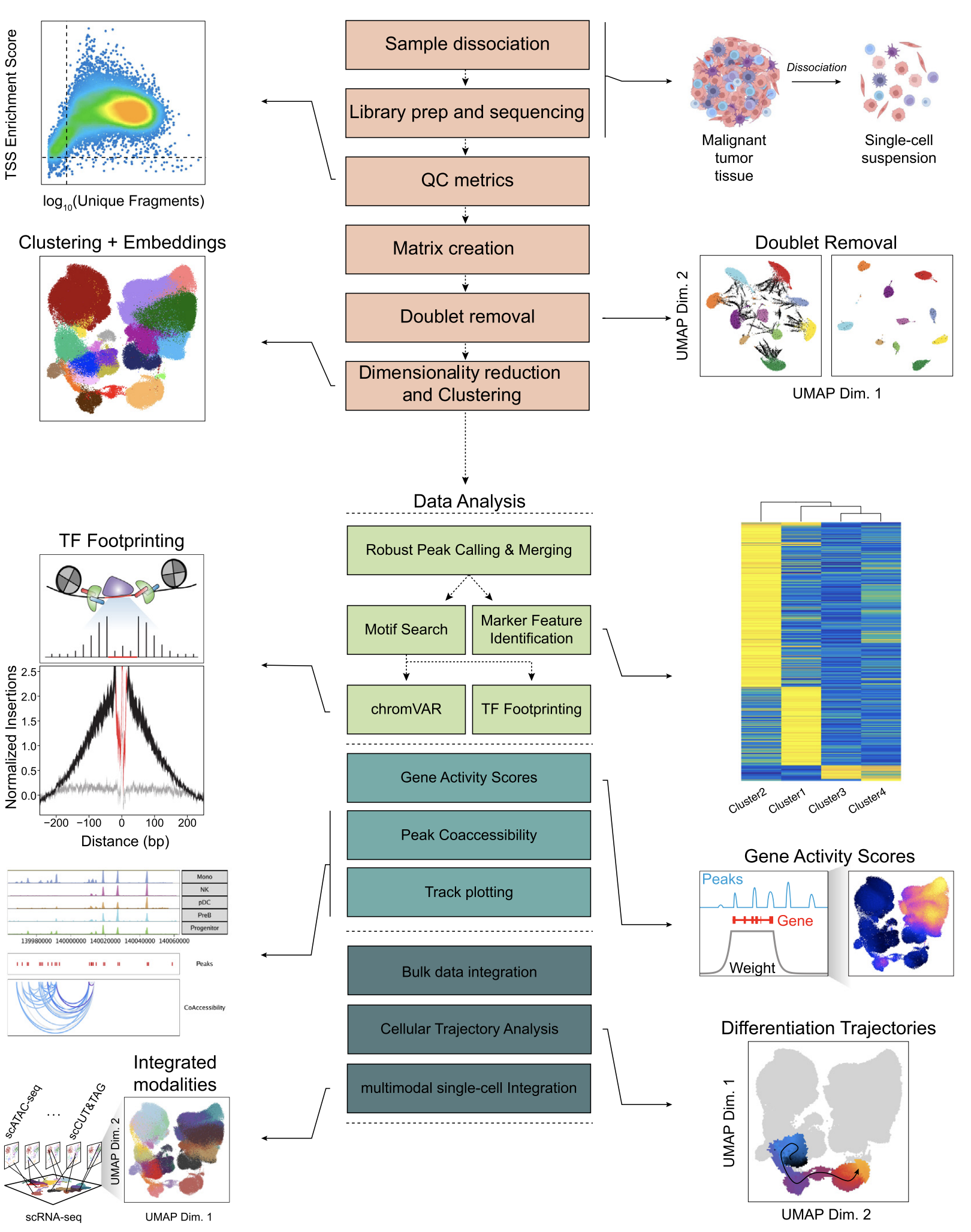
Eyal Metzl-Raz, Samuel H. Kim, Catherine R. Zhang, William J. Greenleaf (2023) Epigenetic Cancer Therapy (book chapter)
Challenges for single-cell epigenetic analysis
Recent advances in single-cell technologies have enabled the profiling of epigenetic landscapes of individual cells, opening a powerful new means for understanding regulatory changes that occur in cancer and cancer-adjacent tissues. In this review, we give an overview of single-cell epigenetic measurement technologies, describe how they have been used in cancer-related studies, highlight their associated challenges, and describe practical considerations for single-cell epigenetic analysis—from cell preparation to data analysis. Finally, we delineate open questions in cancer epigenetic research ripe for investigation with single-cell methods and their corresponding analytical challenges.
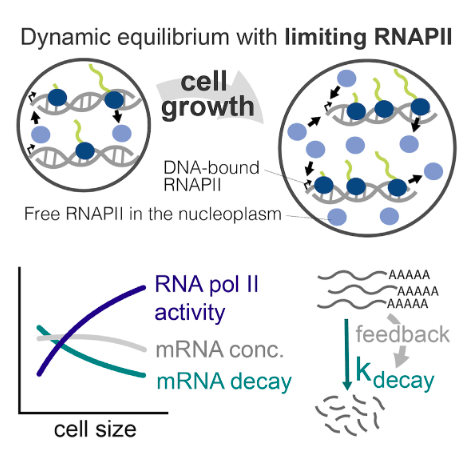
Matthew P Swaffer, Georgi K Marinov, Huan Zheng, Lucas Fuentes Valenzuela, Crystal Yee Tsui, Andrew W Jones, Jessica Greenwood, Anshul Kundaje, William J Greenleaf, Rodrigo Reyes-Lamothe, Jan M Skotheim (2023). Cell.
RNA polymerase II dynamics and mRNA stability feedback scale mRNA amounts with cell size
A fundamental feature of cellular growth is that total protein and RNA amounts increase with cell size to keep concentrations approximately constant. A key component of this is that global transcription rates increase in larger cells. Here, we identify RNA polymerase II (RNAPII) as the limiting factor scaling mRNA transcription with cell size in budding yeast, as transcription is highly sensitive to the dosage of RNAPII but not to other components of the transcriptional machinery. Our experiments support a dynamic equilibrium model where global RNAPII transcription at a given size is set by the mass action recruitment kinetics of unengaged nucleoplasmic RNAPII to the genome. However, this only drives a sub-linear increase in transcription with size, which is then partially compensated for by a decrease in mRNA decay rates as cells enlarge. Thus, limiting RNAPII and feedback on mRNA stability work in concert to scale mRNA amounts with cell size.
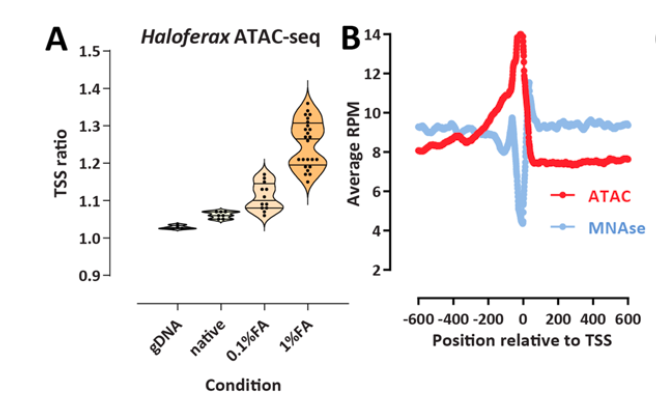
Georgi K Marinov, S Tansu Bagdatli, Tong Wu, Chuan He, Anshul Kundaje, William J Greenleaf (2023). Genome Biology.
The chromatin landscape of the euryarchaeon Haloferax volcanii
Archaea, together with Bacteria, represent the two main divisions of life on Earth, with many of the defining characteristics of the more complex eukaryotes tracing their origin to evolutionary innovations first made in their archaeal ancestors. One of the most notable such features is nucleosomal chromatin, although archaeal histones and chromatin differ significantly from those of eukaryotes, not all archaea possess histones and it is not clear if histones are a main packaging component for all that do. Despite increased interest in archaeal chromatin in recent years, its properties have been little studied using genomic tools.
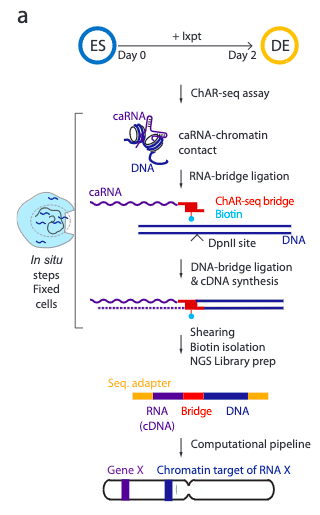
Charles Limouse, Owen K Smith, David Jukam, Kelsey A Fryer, William J Greenleaf, Aaron F Straight (2023). Nature Communications.
Global mapping of RNA-chromatin contacts reveals a proximity-dominated connectivity model for ncRNA-gene interactions
Non-coding RNAs (ncRNAs) are transcribed throughout the genome and provide regulatory inputs to gene expression through their interaction with chromatin. Yet, the genomic targets and functions of most ncRNAs are unknown. Here we use chromatin-associated RNA sequencing (ChAR-seq) to map the global network of ncRNA interactions with chromatin in human embryonic stem cells and the dynamic changes in interactions during differentiation into definitive endoderm. We uncover general principles governing the organization of the RNA-chromatin interactome, demonstrating that nearly all ncRNAs exclusively interact with genes in close three-dimensional proximity to their locus and provide a model predicting the interactome. We uncover RNAs that interact with many loci across the genome and unveil thousands of unannotated RNAs that dynamically interact with chromatin. By relating the dynamics of the interactome to changes in gene expression, we demonstrate that activation or repression of individual genes is unlikely to be controlled by a single ncRNA.
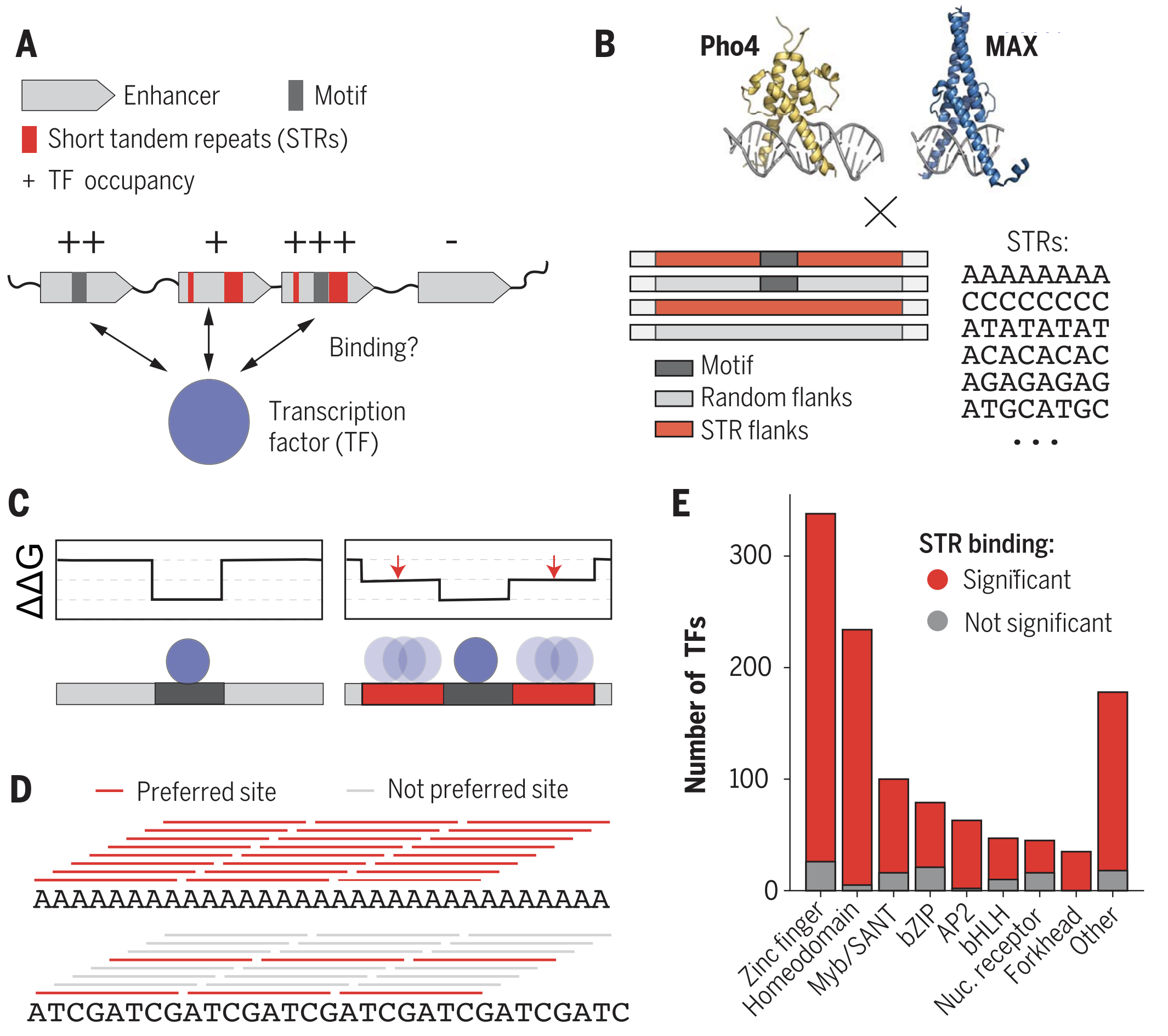
Short tandem repeats bind transcription factors to tune eukaryotic gene expression
Short tandem repeats (STRs) are enriched in eukaryotic cis-regulatory elements and alter gene expression, yet how they regulate transcription remains unknown. We found that STRs modulate transcription factor (TF)-DNA affinities and apparent on-rates by about 70-fold by directly binding TF DNA-binding domains, with energetic impacts exceeding many consensus motif mutations. STRs maximize the number of weakly preferred microstates near target sites, thereby increasing TF density, with impacts well predicted by statistical mechanics. Confirming that STRs also affect TF binding in cells, neural networks trained only on in vivo occupancies predicted effects identical to those observed in vitro. Approximately 90% of TFs preferentially bound STRs that need not resemble known motifs, providing a cis-regulatory mechanism to target TFs to genomic sites.
Shuxiao Chen, Bokai Zhu, Sijia Huang, John W. Hickey, Kevin Z. Lin, Michael Snyder, William J. Greenleaf, Garry P. Nolan, Nancy R. Zhang & Zongming Ma (2023). Nature Biotechnology
Integration of spatial and single-cell data across modalities with weakly linked features
Although single-cell and spatial sequencing methods enable simultaneous measurement of more than one biological modality, no technology can capture all modalities within the same cell. For current data integration methods, the feasibility of cross-modal integration relies on the existence of highly correlated, a priori ‘linked’ features. We describe matching X-modality via fuzzy smoothed embedding (MaxFuse), a cross-modal data integration method that, through iterative coembedding, data smoothing and cell matching, uses all information in each modality to obtain high-quality integration even when features are weakly linked. MaxFuse is modality-agnostic and demonstrates high robustness and accuracy in the weak linkage scenario, achieving 20~70% relative improvement over existing methods under key evaluation metrics on benchmarking datasets. A prototypical example of weak linkage is the integration of spatial proteomic data with single-cell sequencing data. On two example analyses of this type, MaxFuse enabled the spatial consolidation of proteomic, transcriptomic and epigenomic information at single-cell resolution on the same tissue section.

Florian V De Rop, Gert Hulselmans, Chris Flerin, Paula Soler-Vila, Albert Rafels, Valerie Christiaens, Carmen Bravo González-Blas, Domenica Marchese, Ginevra Caratù, Suresh Poovathingal, Orit Rozenblatt-Rosen, Michael Slyper, Wendy Luo, Christoph Muus, Fabiana Duarte, Rojesh Shrestha, S Tansu Bagdatli, M Ryan Corces, Lira Mamanova, Andrew Knights, Kerstin B Meyer, Ryan Mulqueen, Akram Taherinasab, Patrick Maschmeyer, Jörn Pezoldt, Camille Lucie Germaine Lambert, Marta Iglesias, Sebastián R Najle, Zain Y Dossani, Luciano G Martelotto, Zach Burkett, Ronald Lebofsky, José Ignacio Martin-Subero, Satish Pillai, Arnau Sebé-Pedrós, Bart Deplancke, Sarah A Teichmann, Leif S Ludwig, Theodore P Braun, Andrew C Adey, William J Greenleaf, Jason D Buenrostro, Aviv Regev , Stein Aerts, Holger Heyn (2023). Nature Biotechnology.
Systematic benchmarking of single-cell ATAC-sequencing protocols
Single-cell assay for transposase-accessible chromatin by sequencing
(scATAC-seq) has emerged as a powerful tool for dissecting regulatory
landscapes and cellular heterogeneity. However, an exploration of systemic
biases among scATAC-seq technologies has remained absent. In this study,
we benchmark the performance of eight scATAC-seq methods across
47 experiments using human peripheral blood mononuclear cells (PBMCs)
as a reference sample and develop PUMATAC, a universal preprocessing
pipeline, to handle the various sequencing data formats. Our analyses reveal
significant differences in sequencing library complexity and tagmentation
specificity, which impact cell-type annotation, genotype demultiplexing,
peak calling, differential region accessibility and transcription factor motif
enrichment. Our findings underscore the importance of sample extraction,
method selection, data processing and total cost of experiments, offering
valuable guidance for future research. Finally, our data and analysis pipeline
encompasses 169,000 PBMC scATAC-seq profiles and a best practices code
repository for scATAC-seq data analysis, which are freely available to extend
this benchmarking effort to future protocols.
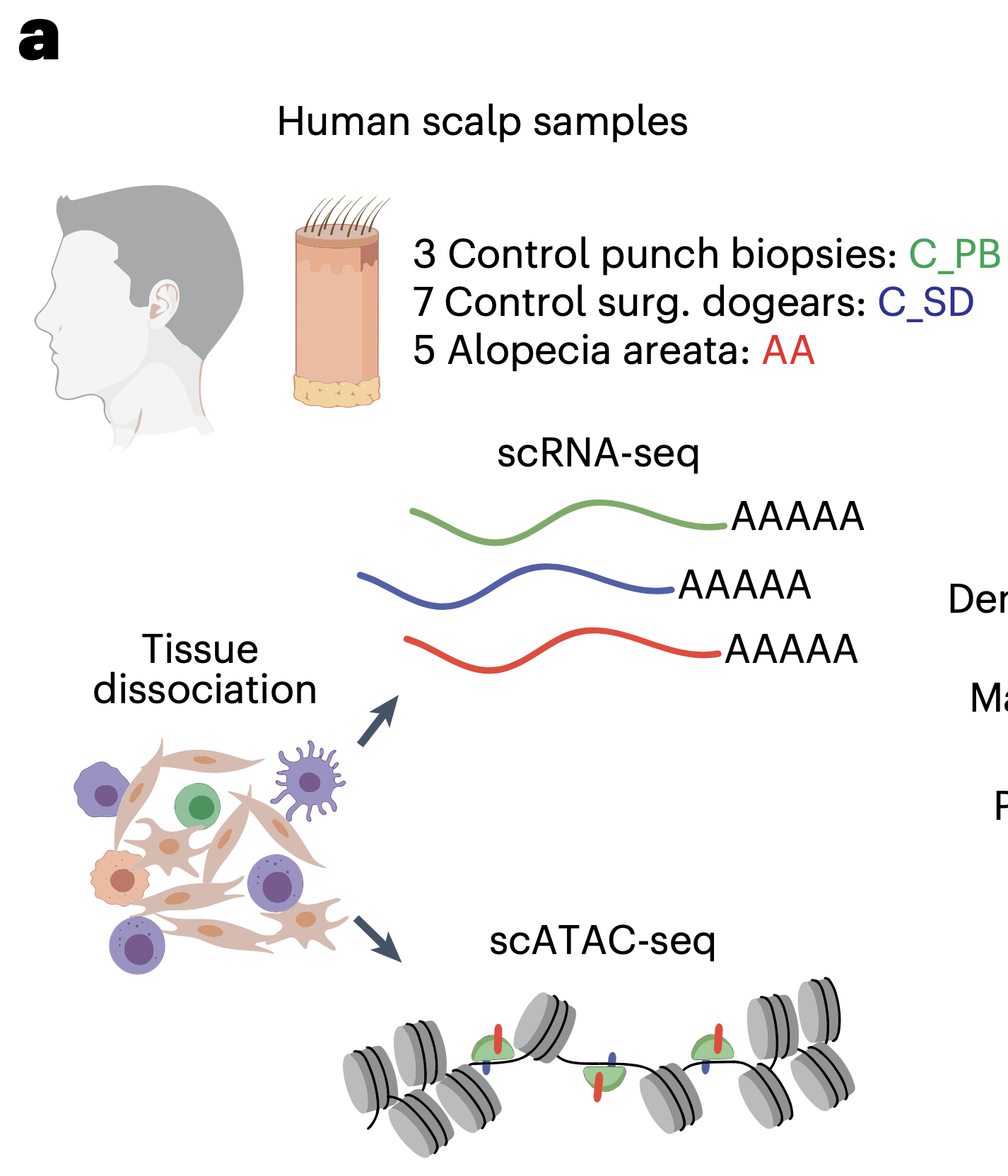
Benjamin Ober-Reynolds, Chen Wang, Justin M. Ko, Eon J. Rios, Sumaira Z. Aasi, Mark M. Davis, Anthony E. Oro, William J. Greenleaf (2023). Nature Genetics.
Integrated single-cell chromatin and transcriptomic analyses of human scalp identify gene-regulatory programs and critical cell types for hair and skin diseases
Genome-wide association studies have identified many loci associated with hair and skin disease, but identification of causal variants requires deciphering of gene-regulatory networks in relevant cell types. We generated matched single-cell chromatin profiles and transcriptomes from scalp tissue from healthy controls and patients with alopecia areata, identifying diverse cell types of the hair follicle niche. By interrogating these datasets at multiple levels of cellular resolution, we infer 50–100% more enhancer–gene links than previous approaches and show that aggregate enhancer accessibility for highly regulated genes predicts expression. We use these gene-regulatory maps to prioritize cell types, genes and causal variants implicated in the pathobiology of androgenetic alopecia (AGA), eczema and other complex traits. AGA genome-wide association studies signals are enriched in dermal papilla regulatory regions, supporting the role of these cells as drivers of AGA pathogenesis. Finally, we train machine learning models to nominate single-nucleotide polymorphisms that affect gene expression through disruption of transcription factor binding, predicting candidate functional single-nucleotide polymorphism for AGA and eczema.
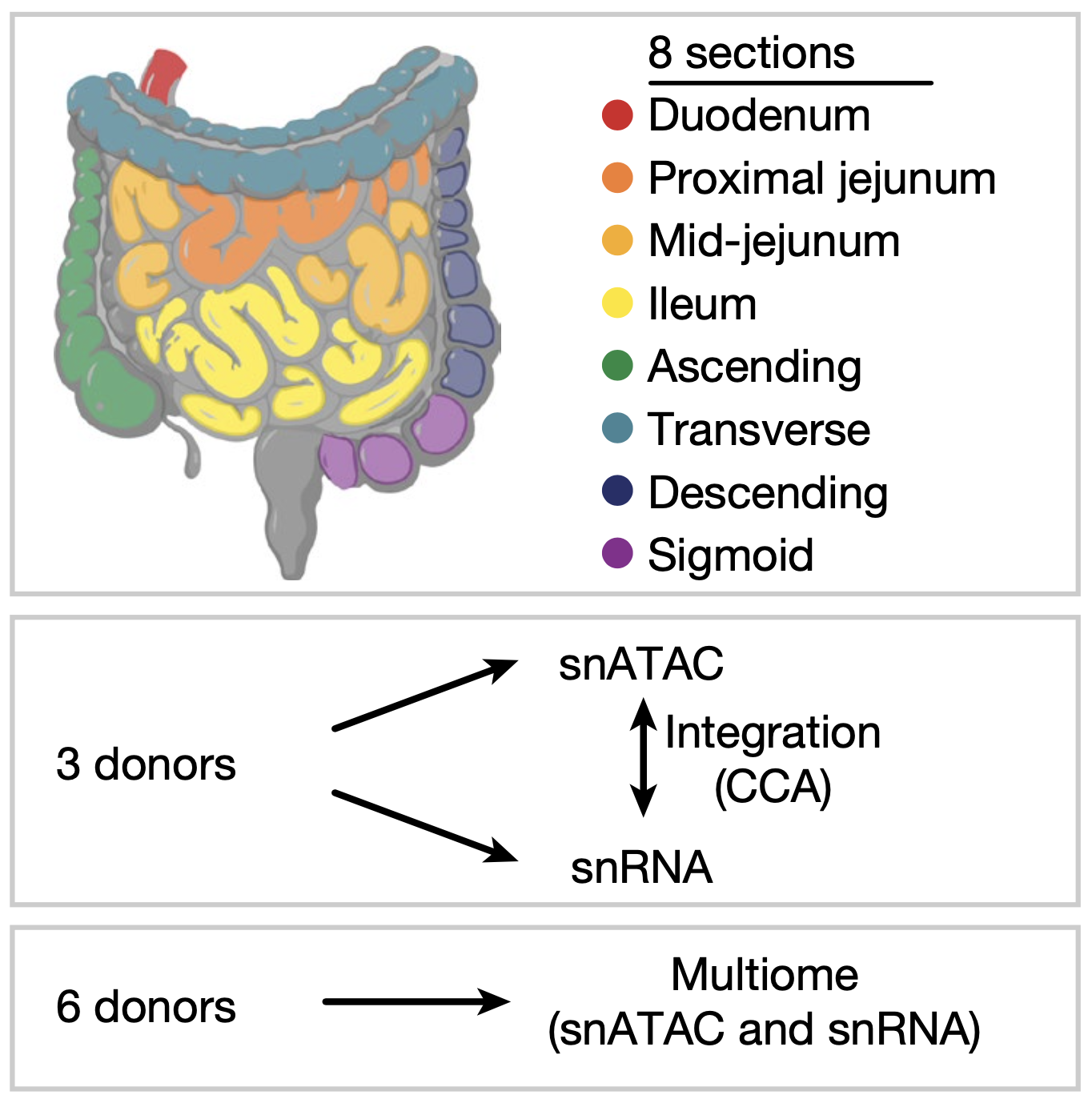
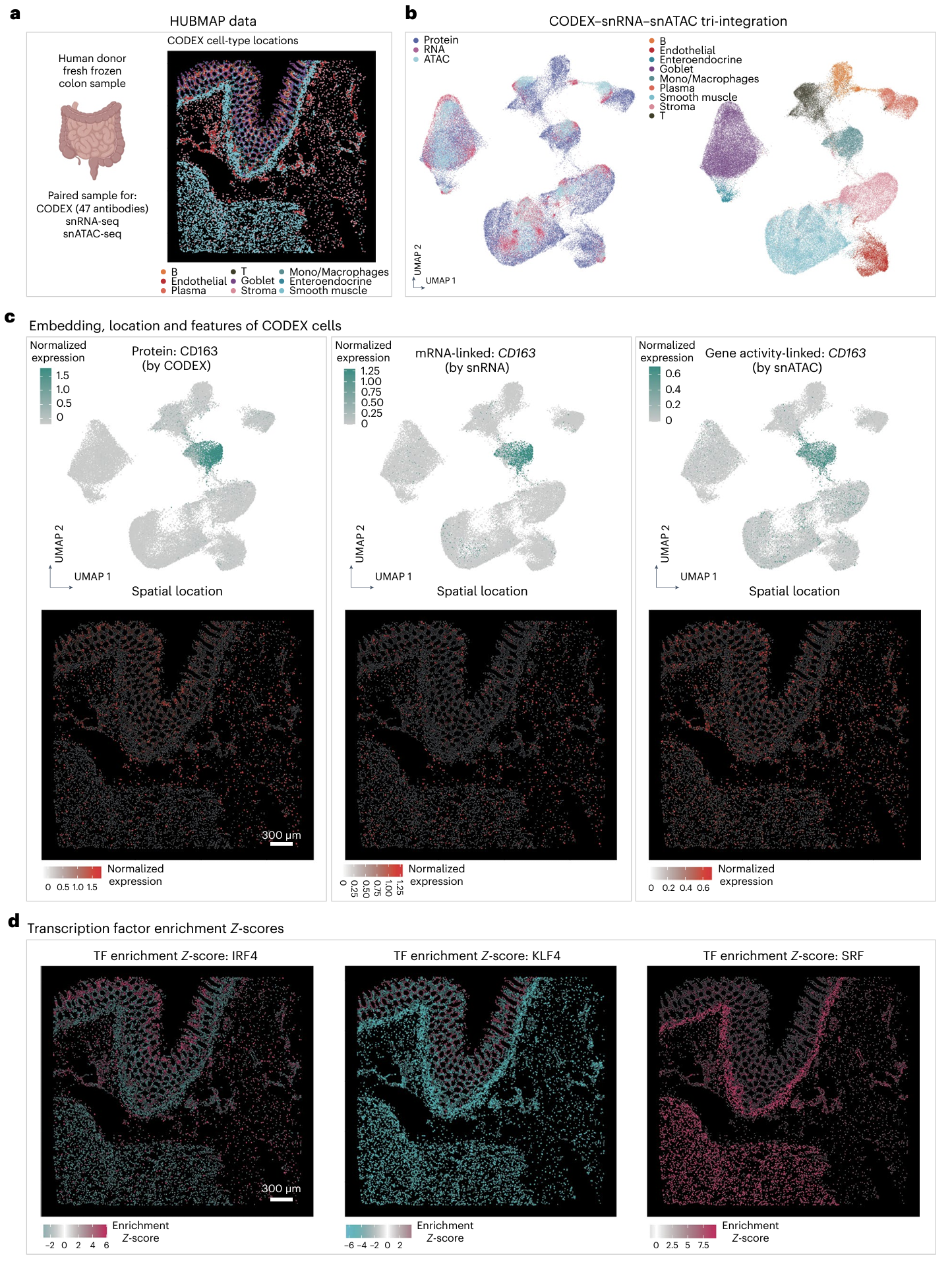
John W. Hickey, Winston R. Becker, Stephanie A. Nevins, Aaron Horning, Almudena Espin Perez, Chenchen Zhu, Bokai Zhu, Bei Wei, Roxanne Chiu, Derek C. Chen, Daniel L. Cotter, Edward D. Esplin, Annika K. Weimer, Chiara Caraccio, Vishal Venkataraaman, Christian M. Schürch, Sarah Black, Maria Brbić, Kaidi Cao, Shuxiao Chen, Weiruo Zhang, Emma Monte, Nancy R. Zhang, Zongming Ma, Jure Leskovec, Zhengyan Zhang, Shin Lin, Teri Longacre, Sylvia K. Plevritis, Yiing Lin, Garry P. Nolan, William J. Greenleaf, Michael Snyder (2023). Nature.
Organization of the human intestine at single-cell resolution
The intestine is a complex organ that promotes digestion, extracts nutrients, participates in immune surveillance, maintains critical symbiotic relationships with microbiota and affects overall health. The intestine has a length of over nine metres, along which there are differences in structure and function. The localization of individual cell types, cell type development trajectories and detailed cell transcriptional programs probably drive these differences in function. Here, to better understand these differences, we evaluated the organization of single cells using multiplexed imaging and single-nucleus RNA and open chromatin assays across eight different intestinal sites from nine donors. Through systematic analyses, we find cell compositions that differ substantially across regions of the intestine and demonstrate the complexity of epithelial subtypes, and find that the same cell types are organized into distinct neighbourhoods and communities, highlighting distinct immunological niches that are present in the intestine. We also map gene regulatory differences in these cells that are suggestive of a regulatory differentiation cascade, and associate intestinal disease heritability with specific cell types. These results describe the complexity of the cell composition, regulation and organization for this organ, and serve as an important reference map for understanding human biology and disease.
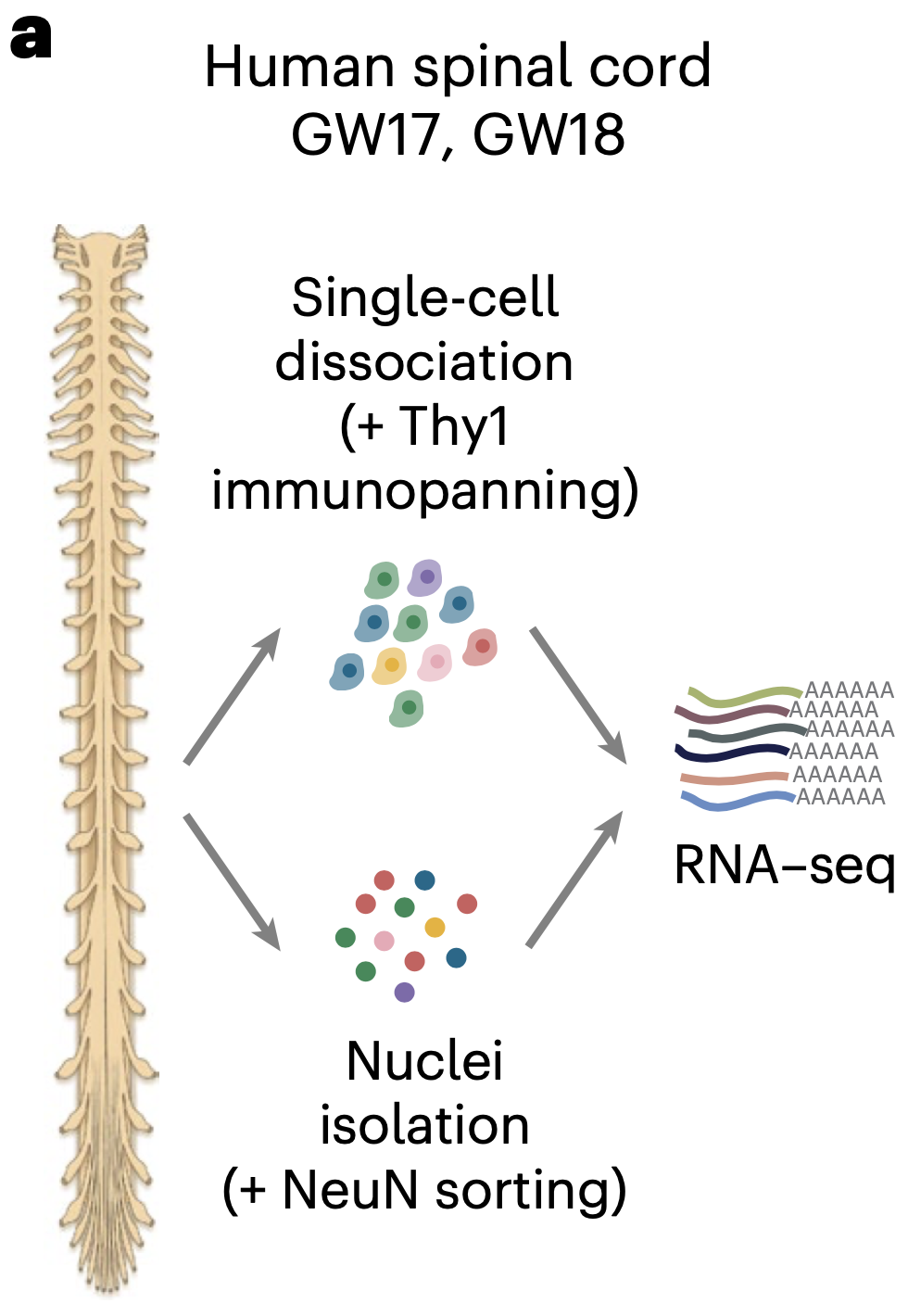
Jimena Andersen, Nicholas Thom, Jennifer L. Shadrach, Xiaoyu Chen, Massimo Mario Onesto, Neal D. Amin, Se-Jin Yoon, Li Li, William J. Greenleaf, Fabian Müller, Anca M. Pașca, Julia A. Kaltschmidt, Sergiu P. Pașca (2023). Nature Neuroscience.
Single-cell transcriptomic landscape of the developing human spinal cord
Understanding spinal cord assembly is essential to elucidate how motor behavior is controlled and how disorders arise. The human spinal cord is exquisitely organized, and this complex organization contributes to the diversity and intricacy of motor behavior and sensory processing. But how this complexity arises at the cellular level in the human spinal cord remains unknown. Here we transcriptomically profiled the midgestation human spinal cord with single-cell resolution and discovered remarkable heterogeneity across and within cell types. Glia displayed diversity related to positional identity along the dorso-ventral and rostro-caudal axes, while astrocytes with specialized transcriptional programs mapped into white and gray matter subtypes. Motor neurons clustered at this stage into groups suggestive of alpha and gamma neurons. We also integrated our data with multiple existing datasets of the developing human spinal cord spanning 22 weeks of gestation to investigate the cell diversity over time. Together with mapping of disease-related genes, this transcriptomic mapping of the developing human spinal cord opens new avenues for interrogating the cellular basis of motor control in humans and guides human stem cell-based models of disease.
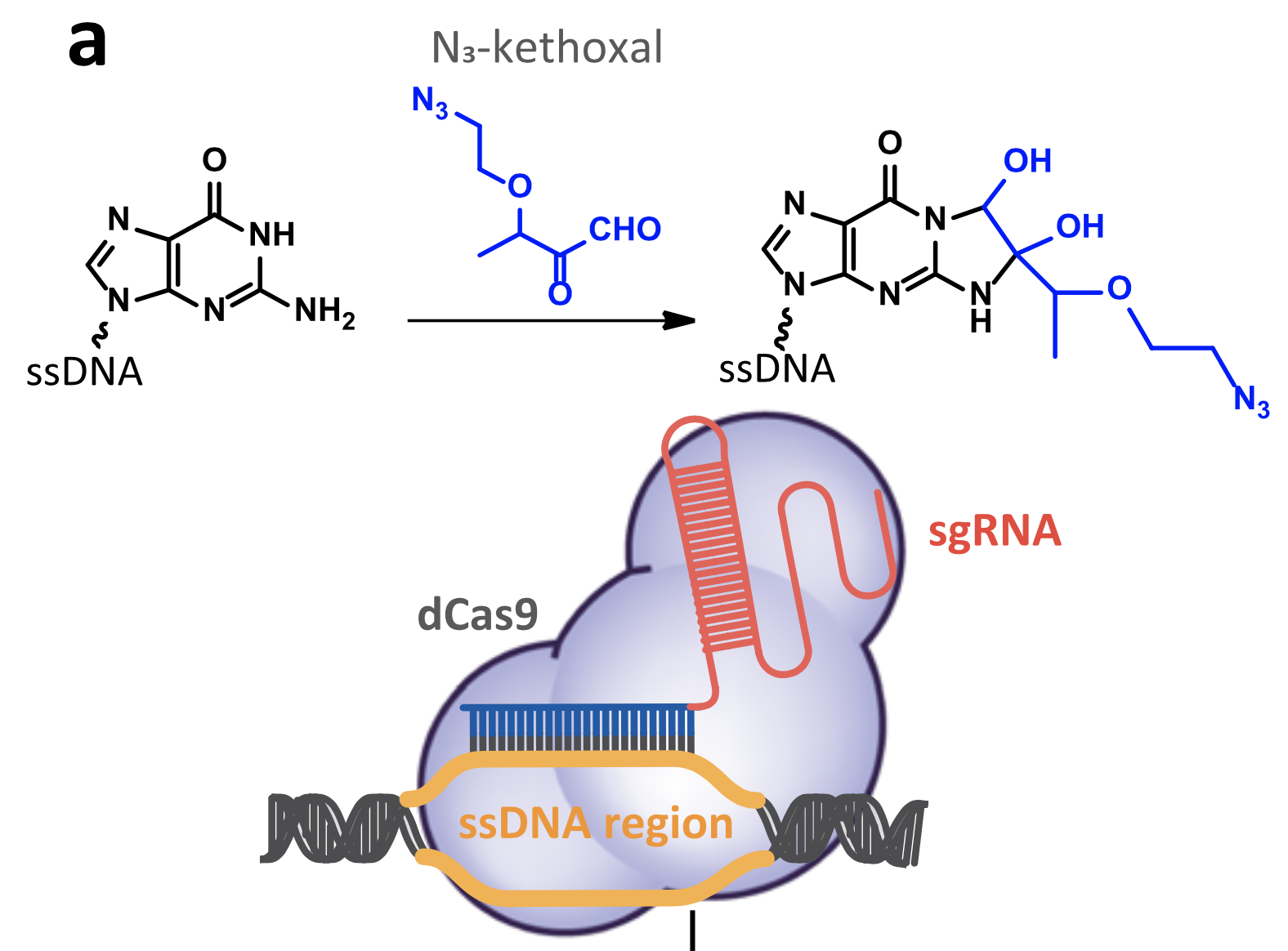
Georgi K. Marinov, Samuel H. Kim, S. Tansu Bagdatli, Soon Il Higashino, Alexandro E. Trevino, Josh Tycko, Tong Wu, Lacramioara Bintu, Michael C. Bassik, Chuan He, Anshul Kundaje, William J. Greenleaf (2023). Genome Biology.
CasKAS: direct profiling of genome-wide dCas9 and Cas9 specificity using ssDNA mapping
Detecting and mitigating off-target activity is critical to the practical application of CRISPR-mediated genome and epigenome editing. While numerous methods have been developed to map Cas9 binding specificity genome-wide, they are generally time-consuming and/or expensive, and not applicable to catalytically dead CRISPR enzymes. We have developed CasKAS, a rapid, inexpensive, and facile assay for identifying off-target CRISPR enzyme binding and cleavage by chemically mapping the unwound single-stranded DNA structures formed upon binding of a sgRNA-loaded Cas9 protein. We demonstrate this method in both in vitro and in vivo contexts.
YeEun Kim, William J Greenleaf, Sean C Bendall (2023). Current Opinion in Immunology.
Systems biology approaches to unravel lymphocyte subsets and function
Single-cell technologies have revealed the extensive heterogeneity and complexity of the immune system. Systems biology approaches in immunology have taken advantage of the high-parameter, high-throughput data and analyzed immune cell types in a ‘bottom-up’ data-driven method. This approach has discovered previously unrecognized cell types and functions. Especially for human immunology, in which experimental manipulations are challenging, systems approach has become a successful means to investigate physiologically relevant contexts. This review focuses on the recent findings in lymphocyte biology, from their development, differentiation into subsets, and heterogeneity in their functions, enabled by these systems approaches. Furthermore, we review examples of the application of findings from systems approach studies and discuss how now to leave the rich dataset in the curse of high dimensionality.

John H. Shin, Steve L. Bonilla, Sarah K. Denny, William J. Greenleaf, Daniel Herschlag (2023). PNAS.
Dissecting the energetic architecture within an RNA tertiary structural motif via high-throughput thermodynamic measurements
Structured RNAs and RNA/protein complexes perform critical cellular functions. They often contain structurally conserved tertiary contact “motifs,” whose occurrence simplifies the RNA folding landscape. Prior studies have focused on the conformational and energetic modularity of intact motifs. Here, we turn to the dissection of one common motif, the 11nt receptor (11ntR), using quantitative analysis of RNA on a massively parallel array to measure the binding of all single and double 11ntR mutants to GAAA and GUAA tetraloops, thereby probing the energetic architecture of the motif. While the 11ntR behaves as a motif, its cooperativity is not absolute. Instead, we uncovered a gradient from high cooperativity amongst base-paired and neighboring residues to additivity between distant residues. As expected, substitutions at residues in direct contact with the GAAA tetraloop resulted in the largest decreases to binding, and energetic penalties of mutations were substantially smaller for binding to the alternate GUAA tetraloop, which lacks tertiary contacts present with the canonical GAAA tetraloop. However, we found that the energetic consequences of base partner substitutions are not, in general, simply described by base pair type or isostericity. We also found exceptions to the previously established stability–abundance relationship for 11ntR sequence variants. These findings of “exceptions to the rule” highlight the power of systematic high-throughput approaches to uncover novel variants for future study in addition to providing an energetic map of a functional RNA.
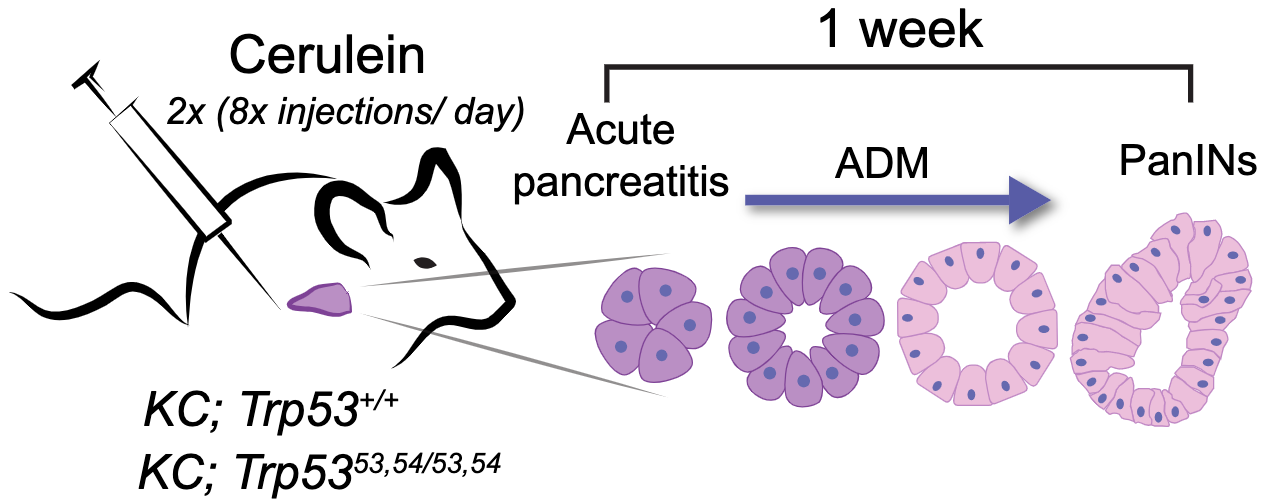
Stephano S. Mello, Brittany M. Flowers, Pawel K. Mazur, James J. Lee, Fabian Müller, Sarah K. Denny, Sofia Ferreira, Kathryn Hanson, Seung K. Kim, William J. Greenleaf, Laura D. Wood, and Laura D. Attardi (2023). PNAS
Multifaceted role for p53 in pancreatic cancer suppression
The vast majority of human pancreatic ductal adenocarcinomas (PDACs) harbor TP53 mutations, underscoring p53’s critical role in PDAC suppression. PDAC can arise when pancreatic acinar cells undergo acinar-to-ductal metaplasia (ADM), giving rise to premalignant pancreatic intraepithelial neoplasias (PanINs), which finally progress to PDAC. The occurrence of TP53 mutations in late-stage PanINs has led to the idea that p53 acts to suppress malignant transformation of PanINs to PDAC. However, the cellular basis for p53 action during PDAC development has not been explored in detail. Here, we leverage a hyperactive p53 variant—p5353,54—which we previously showed is a more robust PDAC suppressor than wild-type p53, to elucidate how p53 acts at the cellular level to dampen PDAC development. Using both inflammation-induced and KRASG12D-driven PDAC models, we find that p5353,54 both limits ADM accumulation and suppresses PanIN cell proliferation and does so more effectively than wild-type p53. Moreover, p5353,54 suppresses KRAS signaling in PanINs and limits effects on the extracellular matrix (ECM) remodeling. While p5353,54 has highlighted these functions, we find that pancreata in wild-type p53 mice similarly show less ADM, as well as reduced PanIN cell proliferation, KRAS signaling, and ECM remodeling relative to Trp53-null mice. We find further that p53 enhances chromatin accessibility at sites controlled by acinar cell identity transcription factors. These findings reveal that p53 acts at multiple stages to suppress PDAC, both by limiting metaplastic transformation of acini and by dampening KRAS signaling in PanINs, thus providing key new understanding of p53 function in PDAC.

Emil Marklund, Yuxi Ke, William J. Greenleaf (2023). Nature Reviews Genetics
High-throughput biochemistry in RNA sequence space: predicting structure and function
RNAs are central to fundamental biological processes in all known organisms. The set of possible intramolecular interactions of RNA nucleotides defines the range of alternative structural conformations of a specific RNA that can coexist, and these structures enable functional catalytic properties of RNAs and/or their productive intermolecular interactions with other RNAs or proteins. However, the immense combinatorial space of potential RNA sequences has precluded predictive mapping between RNA sequence and molecular structure and function. Recent advances in high-throughput approaches in vitro have enabled quantitative thermodynamic and kinetic measurements of RNA–RNA and RNA–protein interactions, across hundreds of thousands of sequence variations. In this Review, we explore these techniques, how they can be used to understand RNA function and how they might form the foundations of an accurate model to predict the structure and function of an RNA directly from its nucleotide sequence. The experimental techniques and modelling frameworks discussed here are also highly relevant for the sampling of sequence–structure–function space of DNAs and proteins.
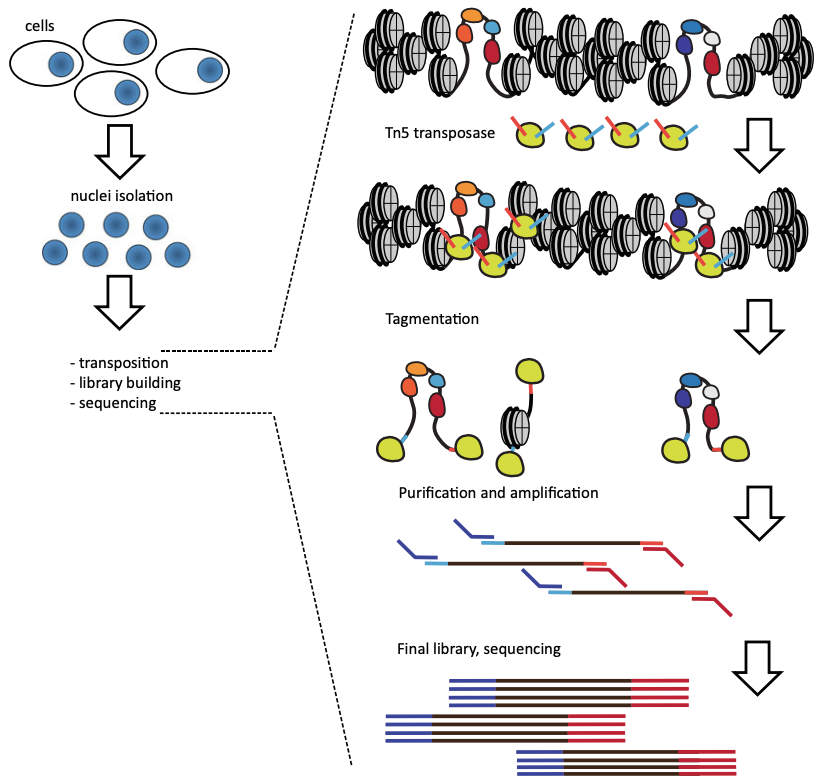
Georgi K. Marinov, Zohar Shipony, Anshul Kundaje, William J. Greenleaf (2023). Methods in Molecular Biology
Genome-Wide Mapping of Active Regulatory Elements Using ATAC-seq
Active cis-regulatory elements (cREs) in eukaryotes are characterized by nucleosomal depletion and, accordingly, higher accessibility. This property has turned out to be immensely useful for identifying cREs genome-wide and tracking their dynamics across different cellular states and is the basis of numerous methods taking advantage of the preferential enzymatic cleavage/labeling of accessible DNA. ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) has emerged as the most versatile and widely adaptable method and has been widely adopted as the standard tool for mapping open chromatin regions. Here, we discuss the current optimal practices and important considerations for carrying out ATAC-seq experiments, primarily in the context of mammalian systems.
Current and future perspectives of single-cell multi-omics technologies in cardiovascular research
Wilson Lek Wen Tan, Wei Qiang Seow, Angela Zhang, Siyeon Rhee, Wing H. Wong, William J. Greenleaf, Joseph C. Wu (2023) Nature Cardiovascular Research
Single-cell technology has become an indispensable tool in cardiovascular research since its first introduction in 2009. Here, we highlight the recent remarkable progress in using single-cell technology to study transcriptomic and epigenetic heterogeneity in cardiac disease and development. We then introduce the key concepts in single-cell multi-omics modalities that apply to cardiovascular research. Lastly, we discuss some of the trending concepts in single-cell technology that are expected to propel cardiovascular research to the next phase of single-cell research.
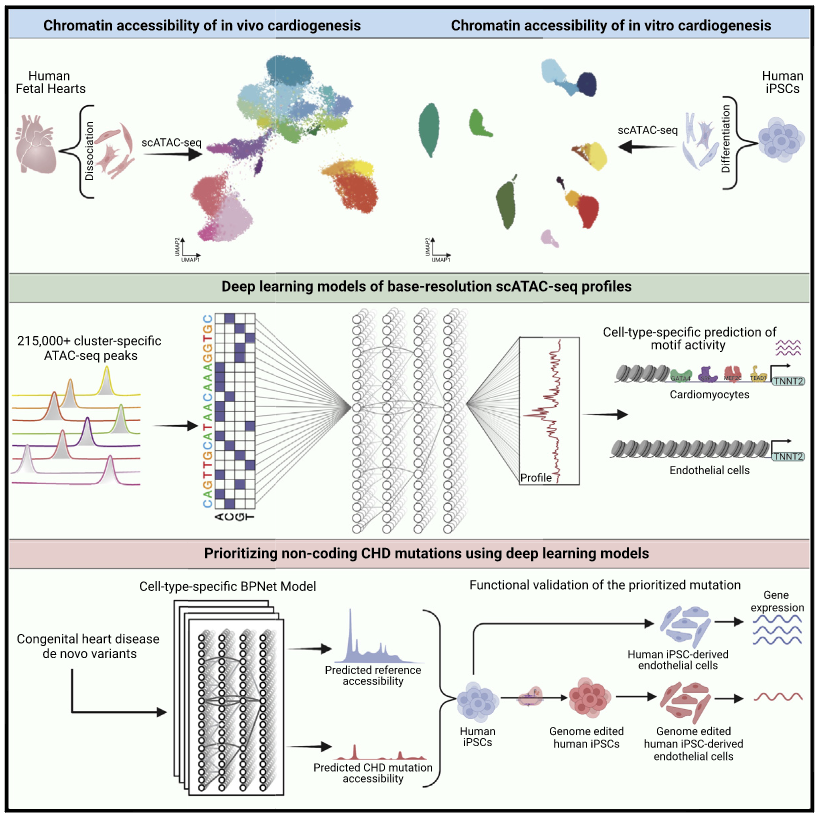
Mohamed Ameen, Laksshman Sundaram, Mengcheng Shen, Abhimanyu Banerjee, Soumya Kundu, Surag Nair, Anna Shcherbina, Mingxia Gu, Kitchener D. Wilson, Avyay Varadarajan, Nirmal Vadgama, Akshay Balsubramani, Joseph C. Wu, Jesse M. Engreitz, Kyle Farh, Ioannis Karakikes, Kevin C. Wang, Thomas Quertermous, William J. Greenleaf, Anshul Kundaje (2022). Cell
Integrative single-cell analysis of cardiogenesis identifies developmental trajectories and non-coding mutations in congenital heart disease
To define the multi-cellular epigenomic and transcriptional landscape of cardiac cellular development, we generated single-cell chromatin accessibility maps of human fetal heart tissues. We identified eight major differentiation trajectories involving primary cardiac cell types, each associated with dynamic transcription factor (TF) activity signatures. We contrasted regulatory landscapes of iPSC-derived cardiac cell types and their in vivo counterparts, which enabled optimization of in vitro differentiation of epicardial cells. Further, we interpreted sequence based deep learning models of cell-type-resolved chromatin accessibility profiles to decipher underlying TF motif lexicons. De novo mutations predicted to affect chromatin accessibility in arterial endothelium were enriched in congenital heart disease (CHD) cases vs. controls. In vitro studies in iPSCs validated the functional impact of identified variation on the predicted developmental cell types. This work thus defines the cell-type-resolved cis-regulatory sequence determinants of heart development and identifies disruption of cell type-specific regulatory elements in CHD.
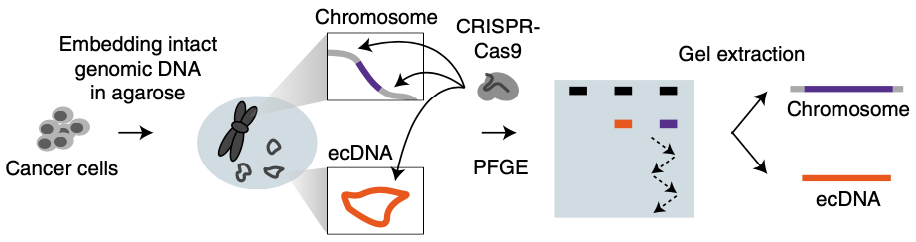
King L. Hung, Jens Luebeck, Siavash R. Dehkordi, Caterina I. Colón, Rui Li, Ivy Tsz-Lo Wong, Ceyda Coruh, Prashanthi Dharanipragada, Shirley H. Lomeli, Natasha E. Weiser, Gatien Moriceau, Xiao Zhang, Chris Bailey, Kathleen E. Houlahan, Wenting Yang, Rocío Chamorro González, Charles Swanton, Christina Curtis, Mariam Jamal-Hanjani, Anton G. Henssen, Julie A. Law, William J. Greenleaf, Roger S. Lo, Paul S. Mischel, Vineet Bafna, Howard Y. Chang (2022) Nature Genetics
Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH
Extrachromosomal DNA (ecDNA) is a common mode of oncogene amplification but is challenging to analyze. Here, we adapt CRISPR-CATCH, in vitro CRISPR-Cas9 treatment and pulsed field gel electrophoresis of agarose-entrapped genomic DNA, previously developed for bacterial chromosome segments, to isolate megabase-sized human ecDNAs. We demonstrate strong enrichment of ecDNA molecules containing EGFR, FGFR2 and MYC from human cancer cells and NRAS ecDNA from human metastatic melanoma with acquired therapeutic resistance. Targeted enrichment of ecDNA versus chromosomal DNA enabled phasing of genetic variants, identified the presence of an EGFRvIII mutation exclusively on ecDNAs and supported an excision model of ecDNA genesis in a glioblastoma model. CRISPR-CATCH followed by nanopore sequencing enabled single-molecule ecDNA methylation profiling and revealed hypomethylation of the EGFR promoter on ecDNAs. We distinguished heterogeneous ecDNA species within the same sample by size and sequence with base-pair resolution and discovered functionally specialized ecDNAs that amplify select enhancers or oncogene-coding sequences.
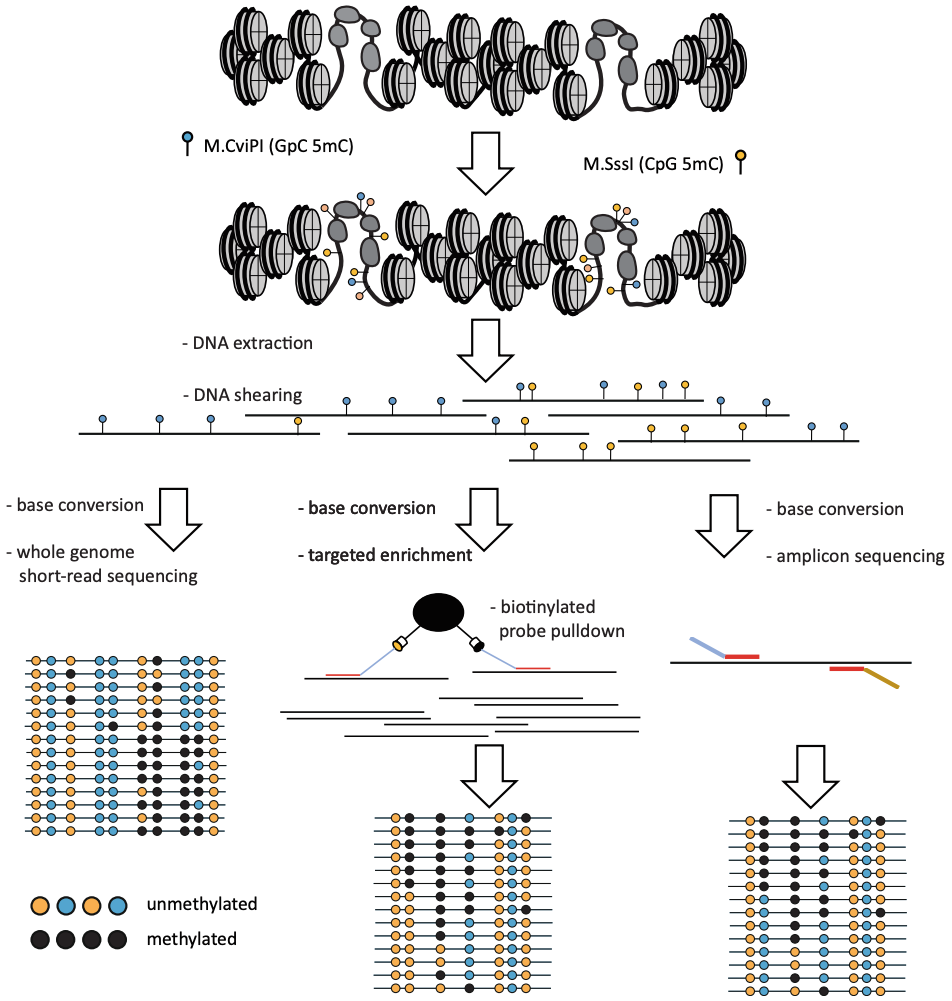
Michaela Hinks, Georgi K. Marinov, Anshul Kundaje, Lacramioara Bintu, William J. Greenleaf (2022). Methods in Molecular Biology
Single-Molecule Mapping of Chromatin Accessibility Using NOMe-seq/dSMF
The bulk of gene expression regulation in most organisms is accomplished through the action of transcription factors (TFs) on cis-regulatory elements (CREs). In eukaryotes, these CREs are generally characterized by nucleosomal depletion and thus higher physical accessibility of DNA. Many methods exploit this property to map regions of high average accessibility, and thus putative active CREs, in bulk. However, these techniques do not provide information about coordinated patterns of accessibility along the same DNA molecule, nor do they map the absolute levels of occupancy/accessibility. SMF (Single-Molecule Footprinting) fills these gaps by leveraging recombinant DNA cytosine methyltransferases (MTase) to mark accessible locations on individual DNA molecules. In this chapter, we discuss current methods and important considerations for performing SMF experiments.
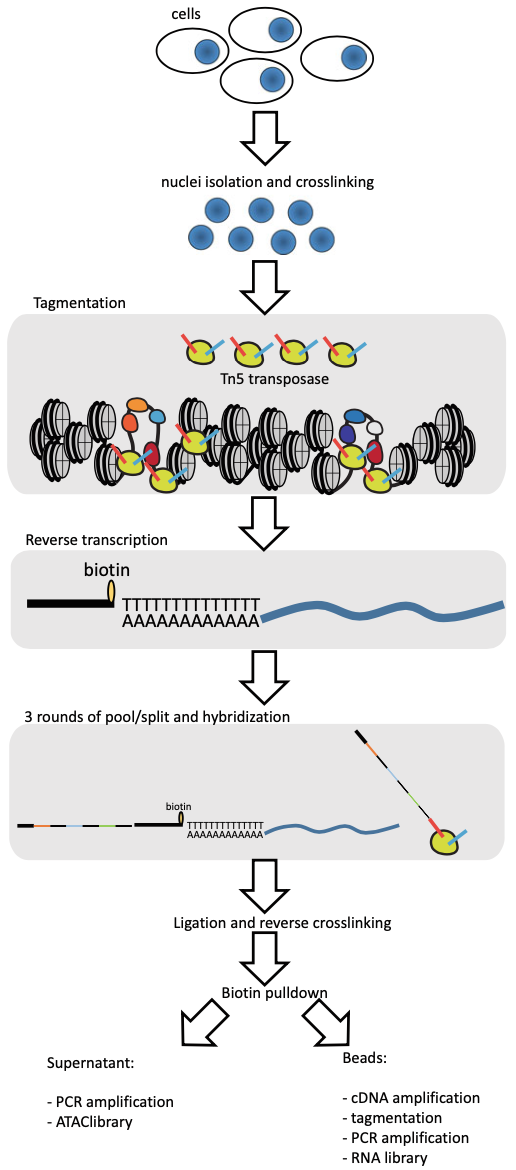
Samuel H. Kim, Georgi K. Marinov, S. Tansu Bagdatli, Soon Il Higashino, Zohar Shipony, Anshul Kundaje, William J. Greenleaf (2022). Methods in Molecular Biology
Simultaneous Single-Cell Profiling of the Transcriptome and Accessible Chromatin Using SHARE-seq
The ability to analyze the transcriptomic and epigenomic states of individual single cells has in recent years transformed our ability to measure and understand biological processes. Recent advancements have focused on increasing sensitivity and throughput to provide richer and deeper biological insights at the cellular level. The next frontier is the development of multiomic methods capable of analyzing multiple features from the same cell, such as the simultaneous measurement of the transcriptome and the chromatin accessibility of candidate regulatory elements. In this chapter, we discuss and describe SHARE-seq (Simultaneous high-throughput ATAC, and RNA expression with sequencing) for carrying out simultaneous chromatin accessibility and transcriptome measurements in single cells, together with the experimental and analytical considerations for achieving optimal results.
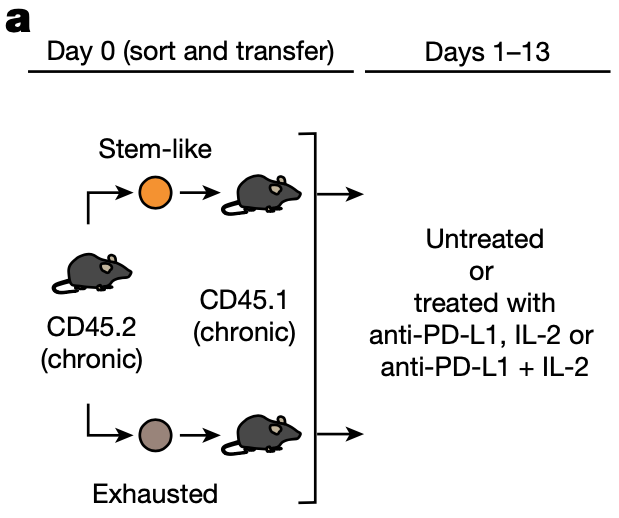
Masao Hashimoto, Koichi Araki, Maria A. Cardenas, Peng Li, Rohit R. Jadhav, Haydn T. Kissick, William H. Hudson, Donald J. McGuire, Rebecca C. Obeng, Andreas Wieland, Judong Lee, Daniel T. McManus, James L. Ross, Se Jin Im, Junghwa Lee, Jian-Xin Lin, Bin Hu, Erin E. West, Christopher D. Scharer, Gordon J. Freeman, Arlene H. Sharpe, Suresh S. Ramalingam, Alex Pellerin, Volker Teichgräber, William J. Greenleaf, Christian Klein, Jorg J. Goronzy, Pablo Umaña, Warren J. Leonard, Kendall A. Smith, Rafi Ahmed (2022) Nature
PD-1 combination therapy with IL-2 modifies CD8+ T cell exhaustion program
Combination therapy with PD-1 blockade and IL-2 is highly effective during chronic lymphocytic choriomeningitis virus infection1. Here we examine the underlying basis for this synergy. We show that PD-1 + IL-2 combination therapy, in contrast to PD-1 monotherapy, substantially changes the differentiation program of the PD-1+TCF1+ stem-like CD8+ T cells and results in the generation of transcriptionally and epigenetically distinct effector CD8+ T cells that resemble highly functional effector CD8+ T cells seen after an acute viral infection. The generation of these qualitatively superior CD8+ T cells that mediate viral control underlies the synergy between PD-1 and IL-2. Our results show that the PD-1+TCF1+ stem-like CD8+ T cells, also referred to as precursors of exhausted CD8+ T cells, are not fate-locked into the exhaustion program and their differentiation trajectory can be changed by IL-2 signals. These virus-specific effector CD8+ T cells emerging from the stem-like CD8+ T cells after combination therapy expressed increased levels of the high-affinity IL-2 trimeric (CD25–CD122–CD132) receptor. This was not seen after PD-1 blockade alone. Finally, we show that CD25 engagement with IL-2 has an important role in the observed synergy between IL-2 cytokine and PD-1 blockade. Either blocking CD25 with an antibody or using a mutated version of IL-2 that does not bind to CD25 but still binds to CD122 and CD132 almost completely abrogated the synergistic effects observed after PD-1 + IL-2 combination therapy. There is considerable interest in PD-1 + IL-2 combination therapy for patients with cancer, and our fundamental studies defining the underlying mechanisms of how IL-2 synergizes with PD-1 blockade should inform these human translational studies.
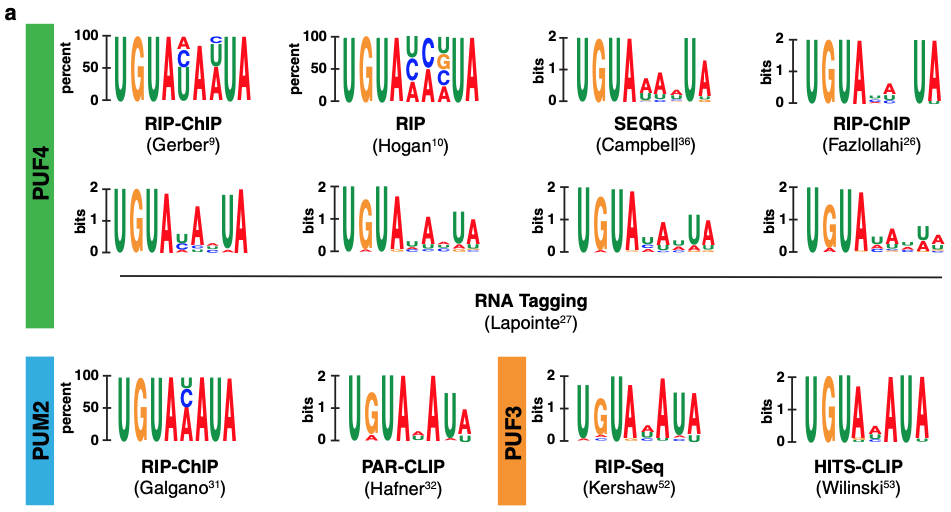
Christoph Sadée, Lauren D. Hagler, Winston R. Becker, Inga Jarmoskaite, Pavanapuresan P. Vaidyanathan, Sarah K. Denny, William J. Greenleaf, Daniel Herschlag (2022) Nature Communications
A comprehensive thermodynamic model for RNA binding by the Saccharomyces cerevisiae Pumilio protein PUF4
Genomic methods have been valuable for identifying RNA-binding proteins (RBPs) and the genes, pathways, and processes they regulate. Nevertheless, standard motif descriptions cannot be used to predict all RNA targets or test quantitative models for cellular interactions and regulation. We present a complete thermodynamic model for RNA binding to the S. cerevisiae Pumilio protein PUF4 derived from direct binding data for 6180 RNAs measured using the RNA on a massively parallel array (RNA-MaP) platform. The PUF4 model is highly similar to that of the related RBPs, human PUM2 and PUM1, with one marked exception: a single favorable site of base flipping for PUF4, such that PUF4 preferentially binds to a non-contiguous series of residues. These results are foundational for developing and testing cellular models of RNA-RBP interactions and function, for engineering RBPs, for understanding the biophysical nature of RBP binding and the evolutionary landscape of RNAs and RBPs.
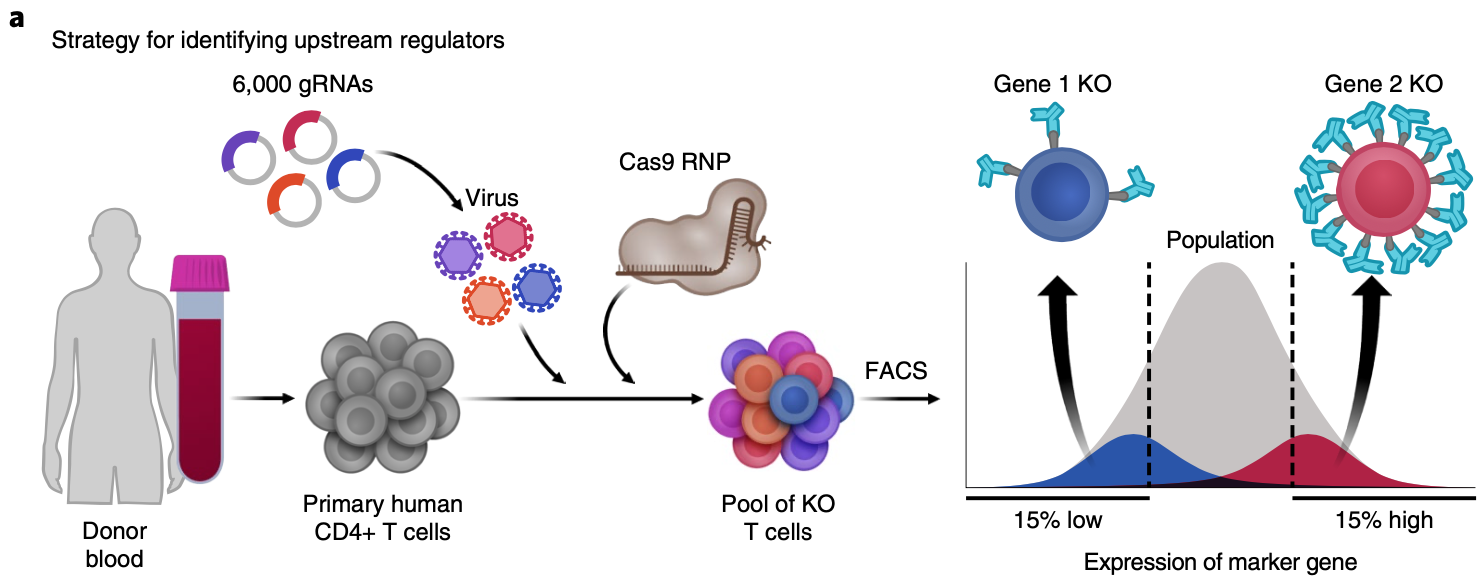
Jacob W. Freimer, Oren Shaked, Sahin Naqvi, Nasa Sinnott-Armstrong, Arwa Kathiria, Christian M. Garrido, Amy F. Chen, Jessica T. Cortez, William J. Greenleaf, Jonathan K. Pritchard, Alexander Marson (2022) Nature Genetics
Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks
Gene regulatory networks ensure that important genes are expressed at precise levels. When gene expression is sufficiently perturbed, it can lead to disease. To understand how gene expression disruptions percolate through a network, we must first map connections between regulatory genes and their downstream targets. However, we lack comprehensive knowledge of the upstream regulators of most genes. Here, we developed an approach for systematic discovery of upstream regulators of critical immune factors—IL2RA, IL-2 and CTLA4—in primary human T cells. Then, we mapped the network of the target genes of these regulators and putative cis-regulatory elements using CRISPR perturbations, RNA-seq and ATAC-seq. These regulators form densely interconnected networks with extensive feedback loops. Furthermore, this network is enriched for immune-associated disease variants and genes. These results provide insight into how immune-associated disease genes are regulated in T cells and broader principles about the structure of human gene regulatory networks.
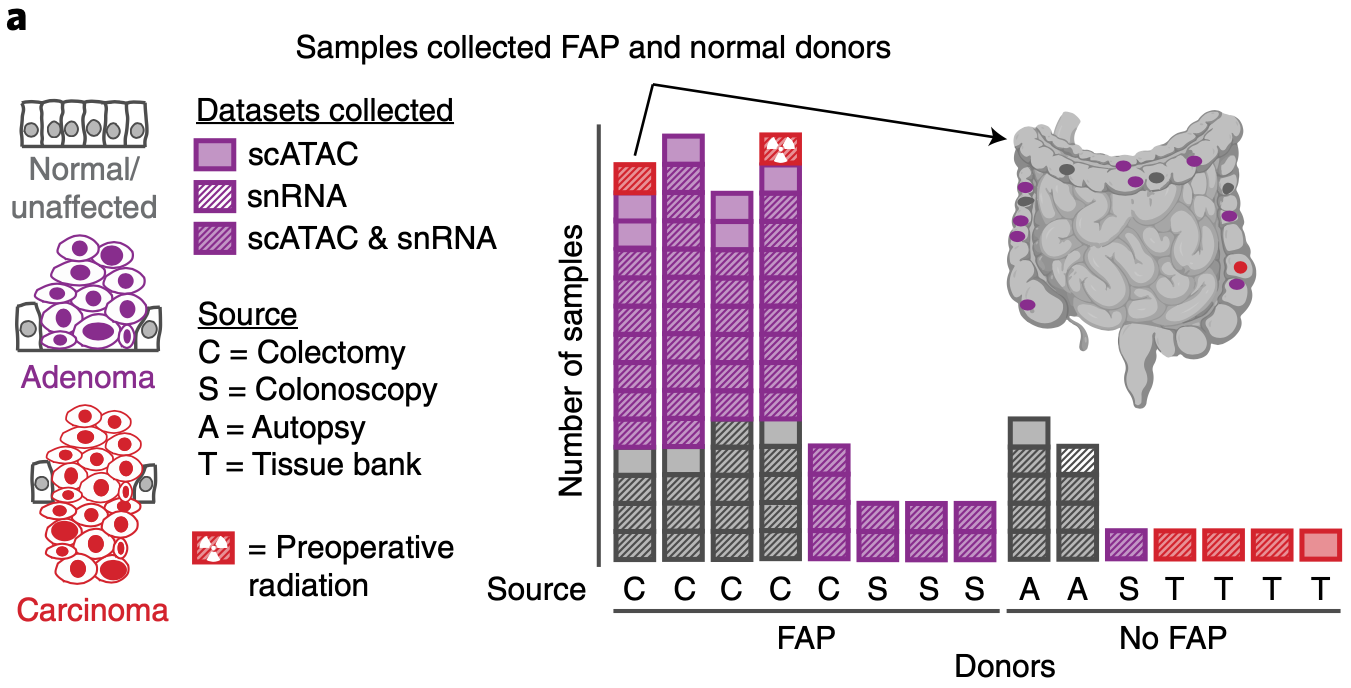
Winston R. Becker, Stephanie A. Nevins, Derek C. Chen, Roxanne Chiu, Aaron M. Horning, Tuhin K. Guha, Rozelle Laquindanum, Meredith Mills, Hassan Chaib, Uri Ladabaum, Teri Longacre, Jeanne Shen, Edward D. Esplin, Anshul Kundaje, James M. Ford, Christina Curtis, Michael P. Snyder, William J. Greenleaf (2022) Nature Genetics
Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer
To chart cell composition and cell state changes that occur during the transformation of healthy colon to precancerous adenomas to colorectal cancer (CRC), we generated single-cell chromatin accessibility profiles and single-cell transcriptomes from 1,000 to 10,000 cells per sample for 48 polyps, 27 normal tissues and 6 CRCs collected from patients with or without germline APC mutations. A large fraction of polyp and CRC cells exhibit a stem-like phenotype, and we define a continuum of epigenetic and transcriptional changes occurring in these stem-like cells as they progress from homeostasis to CRC. Advanced polyps contain increasing numbers of stem-like cells, regulatory T cells and a subtype of pre-cancer-associated fibroblasts. In the cancerous state, we observe T cell exhaustion, RUNX1-regulated cancer-associated fibroblasts and increasing accessibility associated with HNF4A motifs in epithelia. DNA methylation changes in sporadic CRC are strongly anti-correlated with accessibility changes along this continuum, further identifying regulatory markers for molecular staging of polyps.
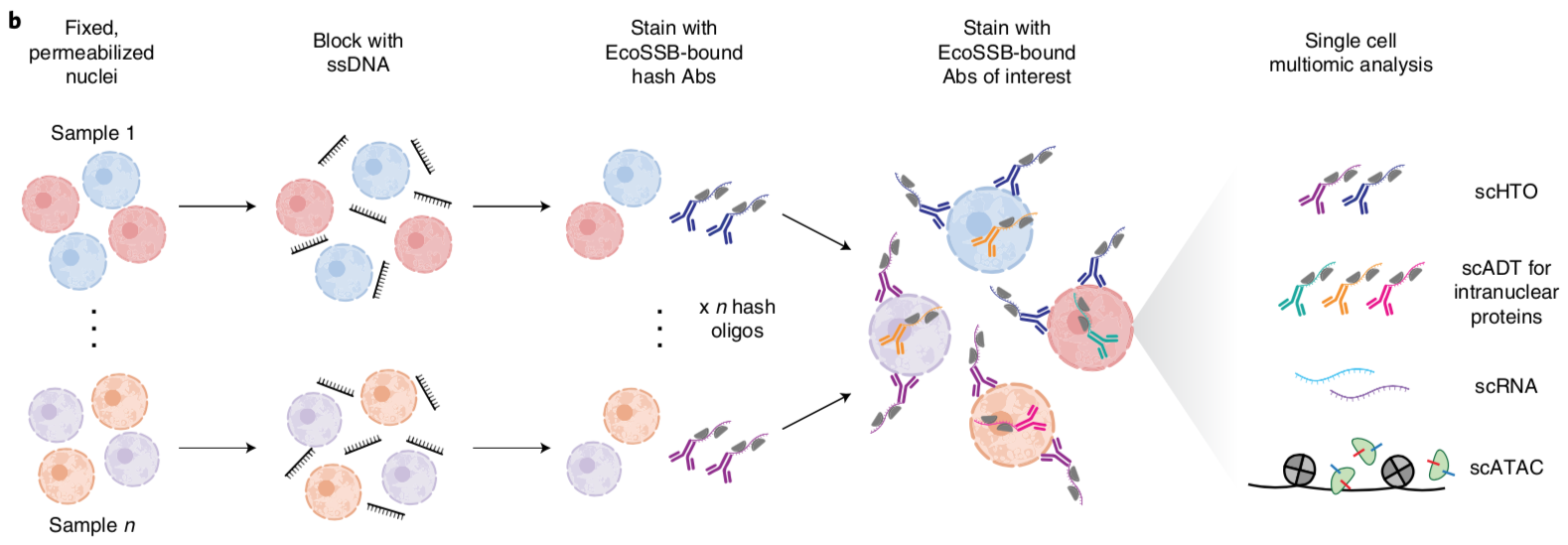
Amy F. Chen, Benjamin Parks, Arwa S. Kathiria, Benjamin Ober-Reynolds, Jorg J. Goronzy, William J. Greenleaf (2022) Nature Methods
NEAT-seq: simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells
In this work, we describe NEAT-seq (sequencing of nuclear protein epitope abundance, chromatin accessibility and the transcriptome in single cells), enabling interrogation of regulatory mechanisms spanning the central dogma. We apply this technique to profile CD4 memory T cells using a panel of master transcription factors (TFs) that drive T cell subsets and identify examples of TFs with regulatory activity gated by transcription, translation and regulation of chromatin binding. We also link a noncoding genome-wide association study single-nucleotide polymorphism (SNP) within a GATA motif to a putative target gene, using NEAT-seq data to internally validate SNP impact on GATA3 regulation.
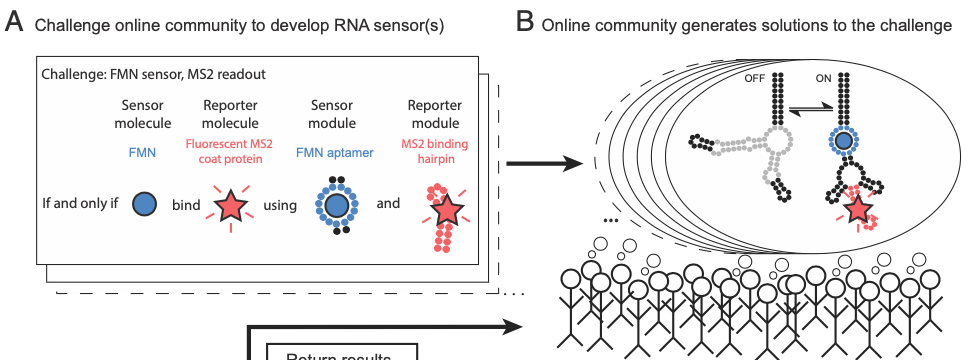
Johan O. L. Andreasson, Michael R. Gotrik, Michelle J. Wu, Hannah K. Wayment-Steele, Wipapat Kladwang, Fernando Portela, Roger Wellington-Oguri, Eterna Participants, Rhiju Das, William J. Greenleaf (2022) PNAS
Crowdsourced RNA design discovers diverse, reversible, efficient, self-contained molecular switches
Internet-based scientific communities promise a means to apply distributed, diverse human intelligence toward previously intractable scientific problems. However, current implementations have not allowed communities to propose experiments to test all emerging hypotheses at scale or to modify hypotheses in response to experiments. We report high-throughput methods for molecular characterization of nucleic acids that enable the large-scale video game–based crowdsourcing of RNA sensor design, followed by high-throughput functional characterization. Iterative design testing of thousands of crowdsourced RNA sensor designs produced near–thermodynamically optimal and reversible RNA switches that act as self-contained molecular sensors and couple five distinct small molecule inputs to three distinct protein binding and fluorogenic outputs. This work suggests a paradigm for widely distributed experimental bioscience.
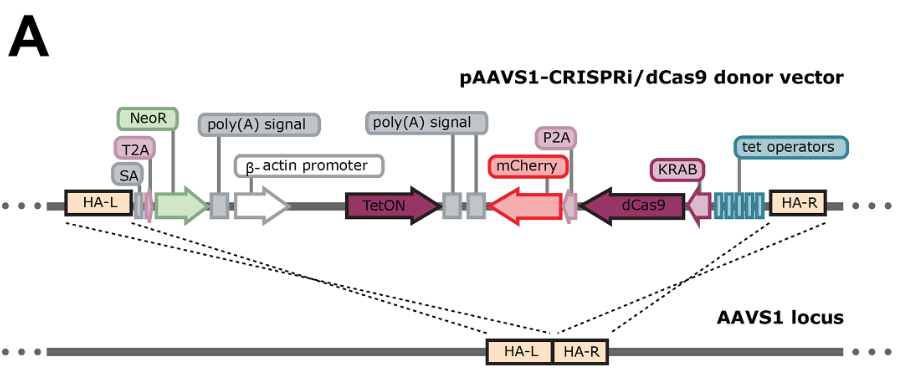
Eyal Metzl-Raz, Nike Bharucha, Jennifer Arthur Ataam, Alexandra A. Gavidia, William J. Greenleaf, Ioannis Karakikes (2022) Stem Cell Research
Generation of a dual edited human induced pluripotent stem cell Myl7-GFP reporter line with inducible CRISPRi/dCas9
Temporal regulation of CRISPRi activity is critical for genetic screens. Here, we present an inducible CRISPRi platform enabling selection of iPSC-derived cardiomyocytes and reversible gene knockdown. We targeted a doxycycline-inducible dCas9-KRAB-mCherry cassette into the AAVS1 locus in an MYL7-mGFP reporter iPSC line. A clone with bi-allelic integration displayed minimally leaky CRISPRi activity and strong expression upon addition of doxycycline in iPSCs, iPSC-derived cardiomyocytes, and multilineage differentiated cells. The CRISPRi activity was validated by targeting the MYOCD gene in iPSC cardiomyocytes. In summary, we developed a robust inducible CRISPRi platform to interrogate gene function in human iPSC-derived cardiomyocytes and other cells.
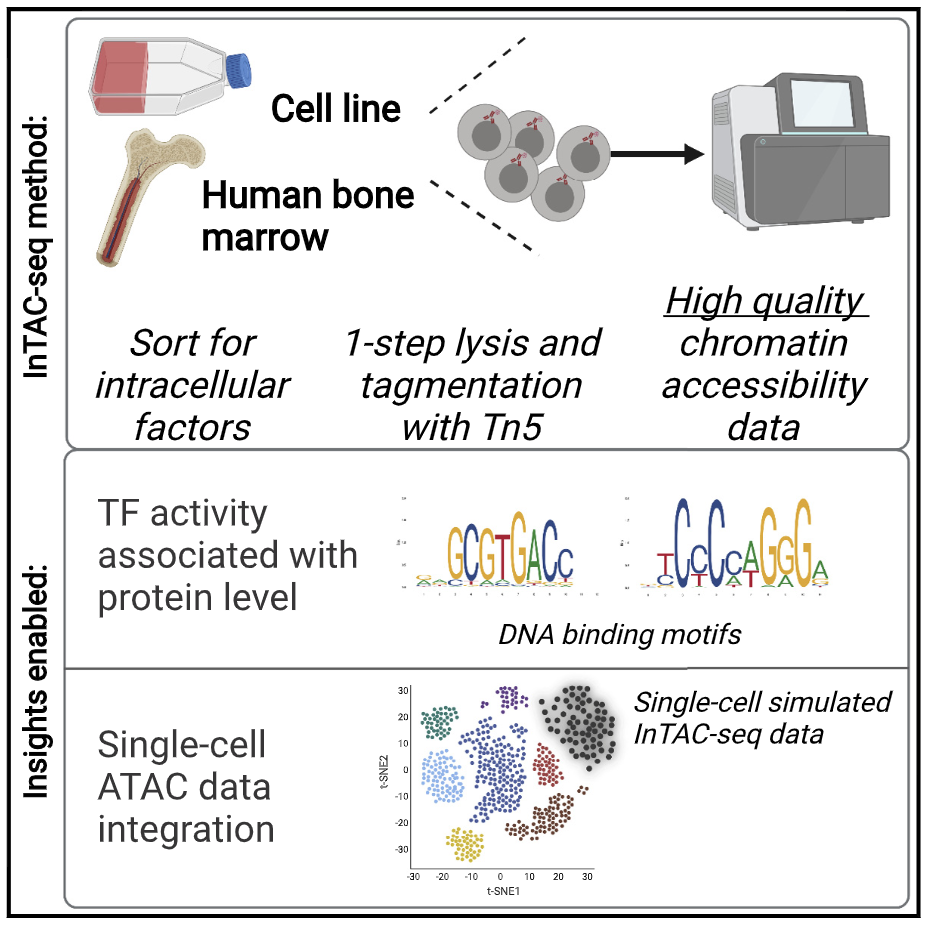
Reema Baskar, Amy F. Chen, Patricia Favaro, Warren Reynolds, Fabian Mueller, Luciene Borges, Sizun Jiang, Hyun Shin Park, Eric T. Kool, William J. Greenleaf, Sean C. Bendall (2022) Cell Reports Methods
Integrating transcription-factor abundance with chromatin accessibility in human erythroid lineage commitment
Master transcription factors (TFs) directly regulate present and future cell states by binding DNA regulatory elements and driving gene-expression programs. Their abundance influences epigenetic priming to different cell fates at the chromatin level, especially in the context of differentiation. In order to link TF protein abundance to changes in TF motif accessibility and open chromatin, we developed InTAC-seq, a method for simultaneous quantification of genome-wide chromatin accessibility and intracellular protein abundance in fixed cells. Our method produces high-quality data and is a cost-effective alternative to single-cell techniques. We showcase our method by purifying bone marrow (BM) progenitor cells based on GATA-1 protein levels and establish high GATA-1-expressing BM cells as both epigenetically and functionally similar to erythroid-committed progenitors.
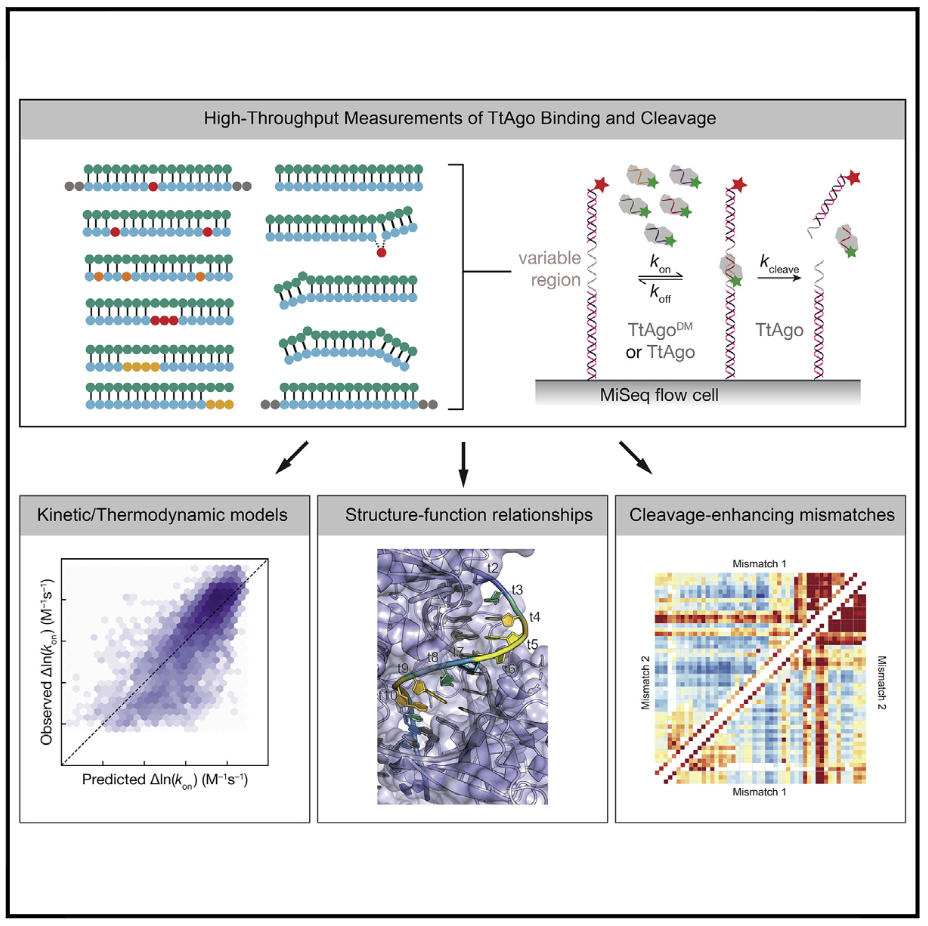
Benjamin Ober-Reynolds, Winston R. Becker, Karina Jouravleva, Samson M. Jolly, Phillip D. Zamore, William J. Greenleaf (2022) Molecular Cell
High-throughput biochemical profiling reveals functional adaptation of a bacterial Argonaute
Argonautes are nucleic acid-guided proteins that perform numerous cellular functions across all domains of life. Little is known about how distinct evolutionary pressures have shaped each Argonaute’s biophysical properties. We applied high-throughput biochemistry to characterize how Thermus thermophilus Argonaute (TtAgo), a DNA-guided DNA endonuclease, finds, binds, and cleaves its targets. We found that TtAgo uses biophysical adaptations similar to those of eukaryotic Argonautes for rapid association but requires more extensive complementarity to achieve high-affinity target binding. Using these data, we constructed models for TtAgo association rates and equilibrium binding affinities that estimate the nucleic acid- and protein-mediated components of the target interaction energies. Finally, we showed that TtAgo cleavage rates vary widely based on the DNA guide, suggesting that only a subset of guides cleaves targets on physiologically relevant timescales.
Georgi K. Marinov, Xinyi Chen, Tong Wu, Chuan He, Arthur R. Grossman, Anshul Kundaje, William James Greenleaf (2022) Genome Biology
The chromatin organization of a chlorarachniophyte nucleomorph genome
Nucleomorphs are remnants of secondary endosymbiotic events between two eukaryote cells wherein the endosymbiont has retained its eukaryotic nucleus. Nucleomorphs have evolved at least twice independently, in chlorarachniophytes and cryptophytes, yet they have converged on a remarkably similar genomic architecture, characterized by the most extreme compression and miniaturization among all known eukaryotic genomes. Previous computational studies have suggested that nucleomorph chromatin likely exhibits a number of divergent features.
Rohit R. Jadhav, Bin Hu, Zhongde Ye, Khushboo Sheth, Xuanying Li, William J .Greenleaf, Cornelia M.Weyand, Jörg J.Goronzy (2022) eBioMedicine
Reduced chromatin accessibility to CD4 T cell super-enhancers encompassing susceptibility loci of rheumatoid arthritis
Rheumatoid arthritis (RA) is an inflammatory disease that manifests as a preclinical stage of systemic autoimmunity followed by chronic progressive synovitis. Disease-associated genetic SNP variants predominantly map to non-coding, regulatory regions of functional importance in CD4 T cells, implicating these cells as key regulators. A better understanding of the epigenome of CD4 T cells holds the promise of providing information on the interaction between genetic susceptibility and exogenous factors.
Georgi K. Marinov, Zohar Shipony, Anshul Kundaje, William J. Greenleaf (2022) Chromatin
Single-Molecule Multikilobase-Scale Profiling of Chromatin Accessibility Using m6A-SMAC-Seq and m6A-CpG-GpC-SMAC-Seq
A hallmark feature of active cis-regulatory elements (CREs) in eukaryotes is their nucleosomal depletion and, accordingly, higher accessibility to enzymatic treatment. This property has been the basis of a number of sequencing-based assays for genome-wide identification and tracking the activity of CREs across different biological conditions, such as DNAse-seq, ATAC-seq, NOMeseq, and others. However, the fragmentation of DNA inherent to many of these assays and the limited read length of short-read sequencing platforms have so far not allowed the simultaneous measurement of the chromatin accessibility state of CREs located distally from each other. The combination of labeling accessible DNA with DNA modifications and nanopore sequencing has made it possible to develop such assays. Here, we provide a detailed protocol for carrying out the SMAC-seq assay (Single-Molecule long-read Accessible Chromatin mapping sequencing), in its m6A-SMAC-seq and m6A-CpG-GpC-SMAC-seq variants, together with methods for data processing and analysis, and discuss key experimental and analytical considerations for working with SMAC-seq datasets.
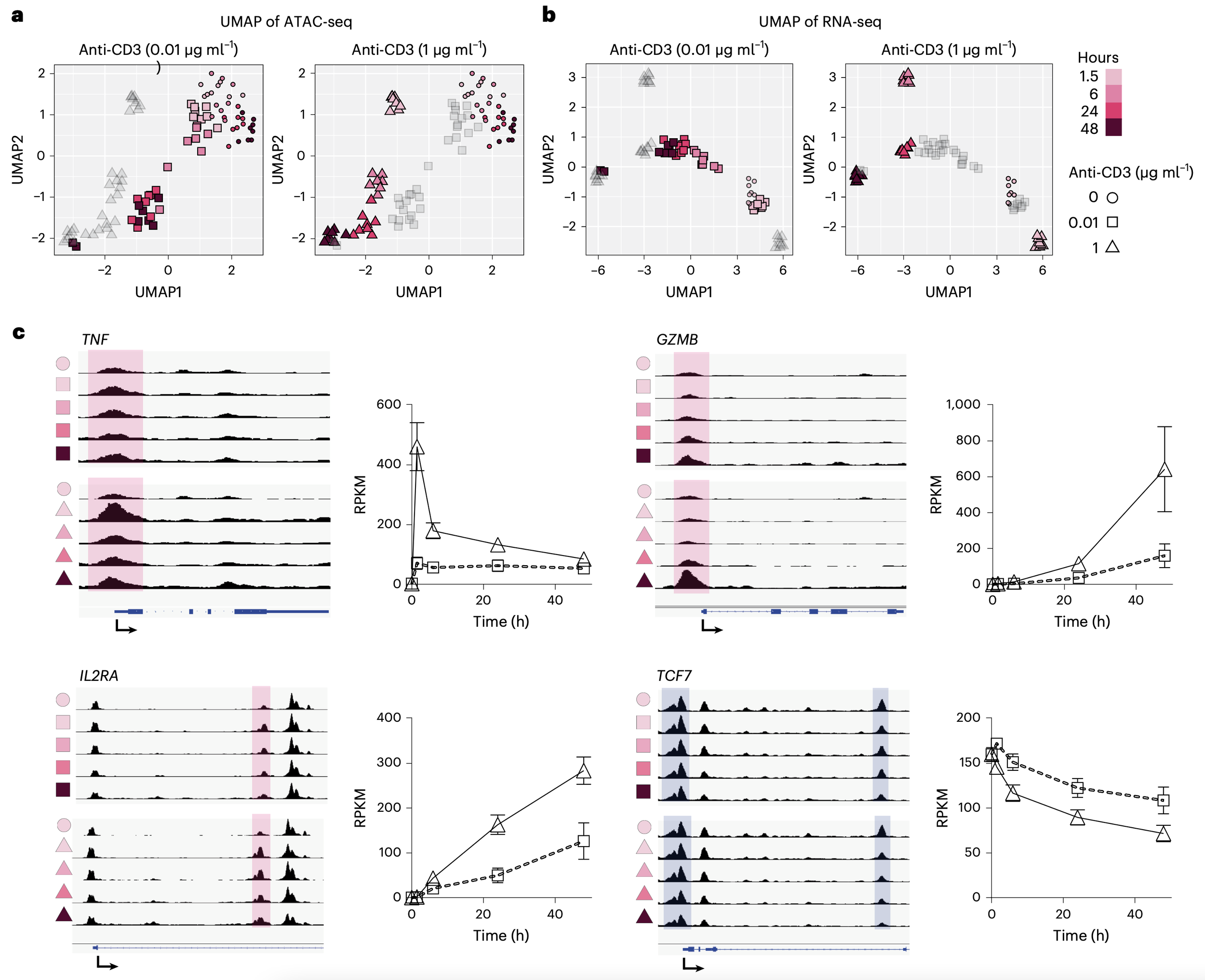
Huimin Zhang, Rohit R. Jadhav, Wenqiang Cao, Isabel N. Goronzy, Tuantuan V. Zhao, Jun Jin, Shozo Ohtsuki, Zhaolan Hu, Jose Morales, William J. Greenleaf, Cornelia M. Weyand, Jörg J. Goronzy (2022) Nature Immunology
Aging-associated HELIOS deficiency in naive CD4+ T cells alters chromatin remodeling and promotes effector cell responses
Immune aging combines cellular defects in adaptive immunity with the activation of pathways causing a low-inflammatory state. Here we examined the influence of age on the kinetic changes in the epigenomic and transcriptional landscape induced by T cell receptor (TCR) stimulation in naive CD4+ T cells. Despite attenuated TCR signaling in older adults, TCR activation accelerated remodeling of the epigenome and induced transcription factor networks favoring effector cell differentiation. We identified increased phosphorylation of STAT5, at least in part due to aberrant IL-2 receptor and lower HELIOS expression, as upstream regulators. Human HELIOS-deficient, naive CD4+ T cells, when transferred into human-synovium-mouse chimeras, infiltrated tissues more efficiently. Inhibition of IL-2 or STAT5 activity in T cell responses of older adults restored the epigenetic response pattern to the one seen in young adults. In summary, reduced HELIOS expression in non-regulatory naive CD4+ T cells in older adults directs T cell fate decisions toward inflammatory effector cells that infiltrate tissue.
Matthew P. Swaffer, Jacob Kim, Devon Chandler-Brown, Maurice Langhinrichs, Georgi K. Marinov, William J. Greenleaf, Anshul Kundaje, Kurt M. Schmoller, Jan M. Skotheim (2021) Molecular Cell
Transcriptional and chromatin-based partitioning mechanisms uncouple protein scaling from cell size
Biosynthesis scales with cell size such that protein concentrations generally remain constant as cells grow. As an exception, synthesis of the cell-cycle inhibitor Whi5 “sub-scales” with cell size so that its concentration is lower in larger cells to promote cell-cycle entry. Here, we find that transcriptional control uncouples Whi5 synthesis from cell size, and we identify histones as the major class of sub-scaling transcripts besides WHI5 by screening for similar genes. Histone synthesis is thereby matched to genome content rather than cell size. Such sub-scaling proteins are challenged by asymmetric cell division because proteins are typically partitioned in proportion to newborn cell volume. To avoid this fate, Whi5 uses chromatin-binding to partition similar protein amounts to each newborn cell regardless of cell size. Disrupting both Whi5 synthesis and chromatin-based partitioning weakens G1 size control. Thus, specific transcriptional and partitioning mechanisms determine protein sub-scaling to control cell size.
Chiung-Ying Chang, Zohar Shipony, Sherry G Lin, Ann Kuo, Xiaochen Xiong, Kyle M Loh, William J Greenleaf, Gerald R Crabtree (2021) Molecular Cell
Increased ACTL6A occupancy within mSWI/SNF chromatin remodelers drives human squamous cell carcinoma
Mammalian SWI/SNF (BAF) chromatin remodelers play dosage-sensitive roles in many human malignancies and neurologic disorders. The gene encoding the BAF subunit actin-like 6a (ACTL6A) is amplified early in the development of many squamous cell carcinomas (SCCs), but its oncogenic role remains unclear. Here we demonstrate that ACTL6A overexpression leads to its stoichiometric assembly into BAF complexes and drives their interaction and engagement with specific regulatory regions in the genome. In normal epithelial cells, ACTL6A was substoichiometric to other BAF subunits. However, increased ACTL6A levels by ectopic expression or in SCC cells led to near saturation of ACTL6A within BAF complexes. Increased ACTL6A occupancy enhanced polycomb opposition genome-wide to activate SCC genes and facilitated the co-dependent loading of BAF and TEAD-YAP complexes on chromatin. Both mechanisms appeared to be critical and function as a molecular AND gate for SCC initiation and maintenance, thereby explaining the specificity of the role of ACTL6A amplification in SCCs.
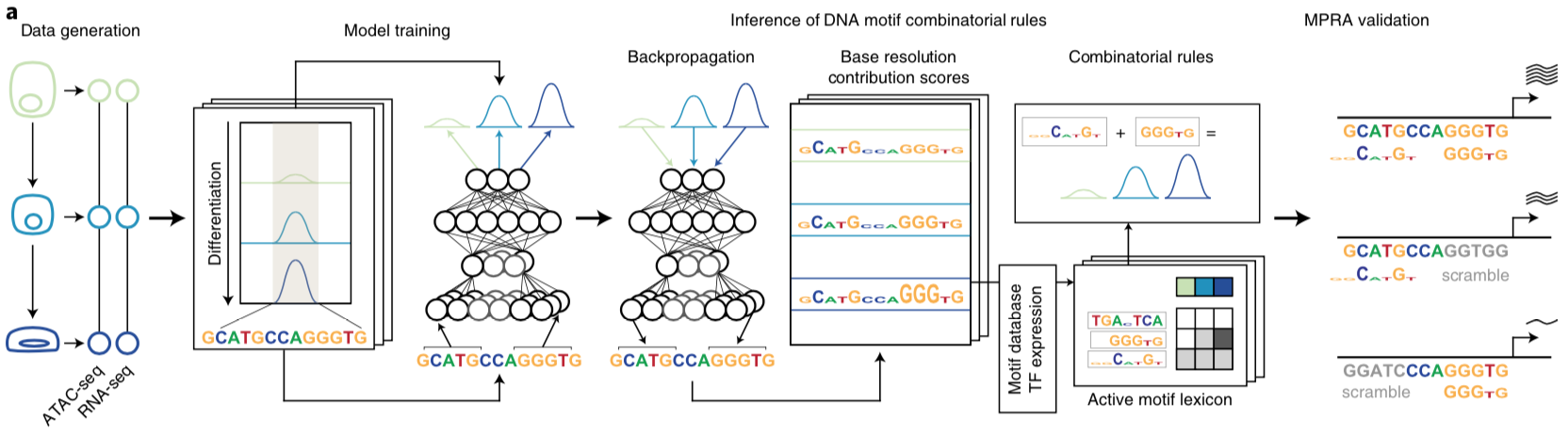
Daniel S. Kim, Viviana I. Risca, David L. Reynolds, James Chappell, Adam J. Rubin, Namyoung Jung, Laura K. H. Donohue, Vanessa Lopez-Pajares, Arwa Kathiria, Minyi Shi, Zhixin Zhao, Harsh Deep, Mahfuza Sharmin, Deepti Rao, Shin Lin, Howard Y. Chang, Michael P. Snyder, William J. Greenleaf, Anshul Kundaje, Paul A. Khavari (2021) Nature Genetics
The dynamic, combinatorial cis-regulatory lexicon of epidermal differentiation
Transcription factors bind DNA sequence motif vocabularies in cis-regulatory elements (CREs) to modulate chromatin state and gene expression during cell state transitions. A quantitative understanding of how motif lexicons influence dynamic regulatory activity has been elusive due to the combinatorial nature of the cis-regulatory code. To address this, we undertook multiomic data profiling of chromatin and expression dynamics across epidermal differentiation to identify 40,103 dynamic CREs associated with 3,609 dynamically expressed genes, then applied an interpretable deep-learning framework to model the cis-regulatory logic of chromatin accessibility. This analysis framework identified cooperative DNA sequence rules in dynamic CREs regulating synchronous gene modules with diverse roles in skin differentiation. Massively parallel reporter assay analysis validated temporal dynamics and cooperative cis-regulatory logic. Variants linked to human polygenic skin disease were enriched in these time-dependent combinatorial motif rules. This integrative approach shows the combinatorial cis-regulatory lexicon of epidermal differentiation and represents a general framework for deciphering the organizational principles of the cis-regulatory code of dynamic gene regulation.
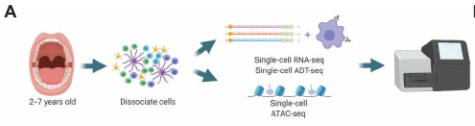
Hamish W. King, Kristen L. Wells, Zohar Shipony, Arwa S. Kathiria, Lisa E. Wagar, Caleb Lareau, Nara Orban, Robson Capasso, Mark M. Davis, Lars M. Steinmetz, Louisa K. James, William J. Greenleaf (2021) Science Immunology
Integrated single-cell transcriptomics and epigenomics reveals strong germinal center–associated etiology of autoimmune risk loci
The germinal center (GC) response is critical for both effective adaptive immunity and establishing peripheral tolerance by limiting autoreactive B cells. Dysfunction in these processes can lead to defective immune responses to infection or contribute to autoimmune disease. To understand the gene regulatory principles underlying the GC response, we generated a single-cell transcriptomic and epigenomic atlas of the human tonsil, a widely studied and representative lymphoid tissue. We characterize diverse immune cell subsets and build a trajectory of dynamic gene expression and transcription factor activity during B cell activation, GC formation, and plasma cell differen-tiation. We subsequently leverage cell type–specific transcriptomic and epigenomic maps to interpret potential regulatory impact of genetic variants implicated in autoimmunity, revealing that many exhibit their greatest reg-ulatory potential in GC-associated cellular populations. These included gene loci linked with known roles in GC biology (IL21, IL21R, IL4R, and BCL6) and transcription factors regulating B cell differentiation (POU2AF1 and HHEX). Together, these analyses provide a powerful new cell type–resolved resource for the interpretation of cellular and genetic causes underpinning autoimmune disease.
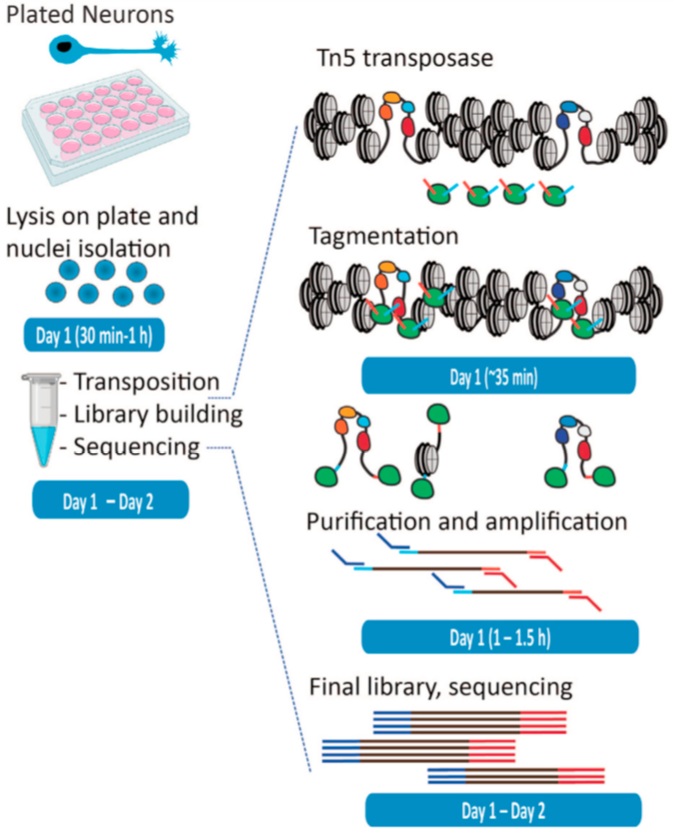
Maya Maor-Nof, Zohar Shipony, Georgi K Marinov, William J Greenleaf, Aaron D Gitler (2021) STAR Protocols
An optimized ATAC-seq protocol for genome-wide mapping of active regulatory elements in primary mouse cortical neurons
ATAC-seq is a versatile, adaptable, and widely adopted technique for mapping open chromatin regions. However, some biological systems, such as primary neurons, present unique challenges to its application. Conventional ATAC-seq would require the dissociation of the primary neurons after plating but dissociating them leads to rapid cell death and major changes in cell state, affecting ATAC-seq results. We have developed this modified ATAC-seq protocol to address this challenge for primary neurons, providing a high-quality and high-resolution accessible chromatin profile.
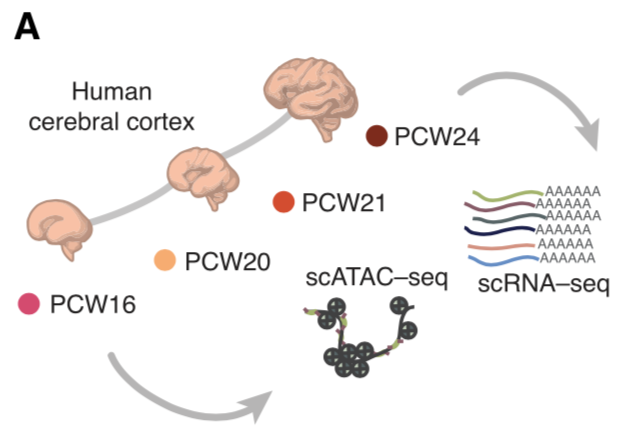
Alexandro E. Trevino, Fabian Muller, Jimena Andersen, Laksshman Sundaram, Arwa Kathiria, Anna Shcherbina, Kyle Farh, Howard Y. Chang, Anca M. Pașca, Anshul Kundaje, Sergiu P. Pașca, William J. Greenleaf (2021) Cell
Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution
Genetic perturbations of cortical development can lead to neurodevelopmental disease, including autism spectrum disorder (ASD). To identify genomic regions crucial to corticogenesis, we mapped the activity of gene-regulatory elements generating a single-cell atlas of gene expression and chromatin accessibility both independently and jointly. This revealed waves of gene regulation by key transcription factors (TFs) across a nearly continuous differentiation trajectory, distinguished the expression programs of glial lineages, and identified lineage-determining TFs that exhibited strong correlation between linked gene-regulatory elements and expression levels. These highly connected genes adopted an active chromatin state in early differentiating cells, consistent with lineage commitment. Base-pair-resolution neural network models identified strong cell-type-specific enrichment of noncoding mutations predicted to be disruptive in a cohort of ASD individuals and identified frequently disrupted TF binding sites. This approach illustrates how cell-type-specific mapping can provide insights into the programs governing human development and disease.
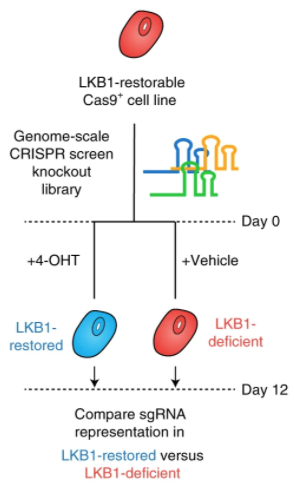
Sarah E. Pierce, Jeffrey M. Granja, M. Ryan Corces, Jennifer J. Brady, Min K. Tsai, Aubrey B. Pierce, Rui Tang, Pauline Chu, David M. Feldser, Howard Y. Chang, Michael C. Bassik, William J. Greenleaf, Monte M. Winslow (2021) Nature Cell Biology
LKB1 inactivation modulates chromatin accessibility to drive metastatic progression
Metastasis is the leading cause of cancer-related deaths and enables cancer cells to compromise organ function by expanding in secondary sites. Since primary tumours and metastases often share the same constellation of driver mutations, the mechanisms that drive their distinct phenotypes are unclear. Here we show that inactivation of the frequently mutated tumour suppressor gene LKB1 (encoding liver kinase B1) has evolving effects throughout the progression of lung cancer, which leads to the differential epigenetic re-programming of early-stage primary tumours compared with late-stage metastases. By integrating genome-scale CRISPR–Cas9 screening with bulk and single-cell multi-omic analyses, we unexpectedly identify LKB1 as a master regulator of chromatin accessibility in lung adenocarcinoma primary tumours. Using an in vivo model of metastatic progression, we further show that loss of LKB1 activates the early endoderm transcription factor SOX17 in metastases and a metastatic-like sub-population of cancer cells within primary tumours. The expression of SOX17 is necessary and sufficient to drive a second wave of epigenetic changes in LKB1-deficient cells that enhances metastatic ability. Overall, our study demonstrates how the downstream effects of an individual driver mutation can change throughout cancer development, with implications for stage-specific therapeutic resistance mechanisms and the gene regulatory underpinnings of metastatic evolution.
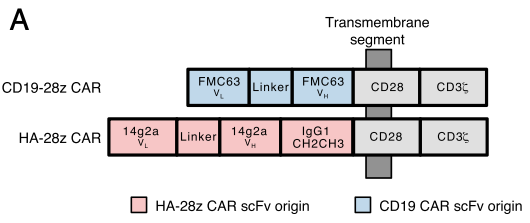
David G. Gennert, Rachel C. Lynn, Jeff M. Granja, Evan W. Weber, Maxwell R. Mumbach, Yang Zhao, Zhana Duren, Elena Sotillo, William J. Greenleaf, Wing H. Wong, Ansuman T. Satpathy, Crystal L. Mackall, Howard Y. Chang. (2021) PNAS
Dynamic chromatin regulatory landscape of human CAR T cell exhaustion
Dysfunction in T cells limits the efficacy of cancer immunotherapy. We profiled the epigenome, transcriptome, and enhancer connectome of exhaustion-prone GD2-targeting HA-28z chimeric antigen receptor (CAR) T cells and control CD19-targeting CAR T cells, which present less exhaustion-inducing tonic signaling, at multiple points during their ex vivo expansion. We found widespread, dynamic changes in chromatin accessibility and three-dimensional (3D) chromosome conformation preceding changes in gene expression, notably at loci proximal to exhaustion-associated genes such as PDCD1, CTLA4, and HAVCR2, and increased DNA motif access for AP-1 family transcription factors, which are known to promote exhaustion. Although T cell exhaustion has been studied in detail in mice, we find that the regulatory networks of T cell exhaustion differ between species and involve distinct loci of accessible chromatin and cis-regulated target genes in human CAR T cell exhaustion. Deletion of exhaustion-specific candidate enhancers of PDCD1 suppress the expression of PD-1 in an in vitro model of T cell dysfunction and in HA-28z CAR T cells, suggesting enhancer editing as a path forward in improving cancer immunotherapy.
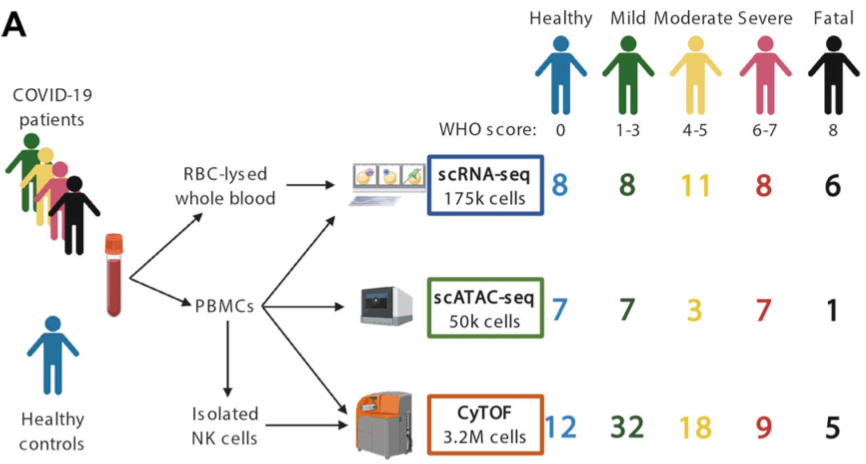
Aaron J. Wilk, Madeline J. Lee, Bei Wei, Benjamin Parks, Ruoxi Pi, Giovanny J. Martínez-Colón, Thanmayi Ranganath, Nancy Q. Zhao, Shalina Taylor, Winston Becker, Stanford COVID-19 Biobank, David Jimenez-Morales, Andra L. Blomkalns, Ruth O’Hara, Euan A. Ashley, Kari C. Nadeau, Samuel Yang, Susan Holmes, Marlene Rabinovitch, Angela J. Rogers, William J. Greenleaf, Catherine A. Blish (2021) Journal of Experimental Medicine
Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19
Our understanding of protective versus pathological immune responses to SARS-CoV-2, the virus that causes coronavirus disease 2019 (COVID-19), is limited by inadequate profiling of patients at the extremes of the disease severity spectrum. Here, we performed multi-omic single-cell immune profiling of 64 COVID-19 patients across the full range of disease severity, from outpatients with mild disease to fatal cases. Our transcriptomic, epigenomic, and proteomic analyses revealed widespread dysfunction of peripheral innate immunity in severe and fatal COVID-19, including prominent hyperactivation signatures in neutrophils and NK cells. We also identified chromatin accessibility changes at NF-κB binding sites within cytokine gene loci as a potential mechanism for the striking lack of pro-inflammatory cytokine production observed in monocytes in severe and fatal COVID-19. We further demonstrated that emergency myelopoiesis is a prominent feature of fatal COVID-19. Collectively, our results reveal disease severity–associated immune phenotypes in COVID-19 and identify pathogenesis-associated pathways that are potential targets for therapeutic intervention.
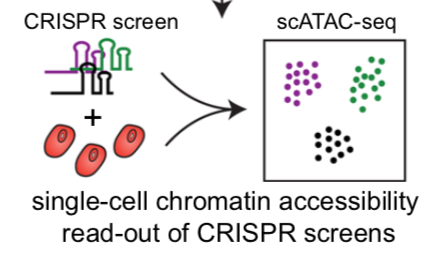
Sarah E. Pierce, Jeffrey M. Granja, William J. Greenleaf (2021) Nature Communications.
High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer
Chromatin accessibility profiling can identify putative regulatory regions genome wide; however, pooled single-cell methods for assessing the effects of regulatory perturbations on accessibility are limited. Here, we report a modified droplet-based single-cell ATAC-seq protocol for perturbing and evaluating dynamic single-cell epigenetic states. This method (Spear-ATAC) enables simultaneous read-out of chromatin accessibility profiles and integrated sgRNA spacer sequences from thousands of individual cells at once. Spear-ATAC profiling of 104,592 cells representing 414 sgRNA knock-down populations reveals the temporal dynamics of epigenetic responses to regulatory perturbations in cancer cells and the associations between transcription factor binding profiles.
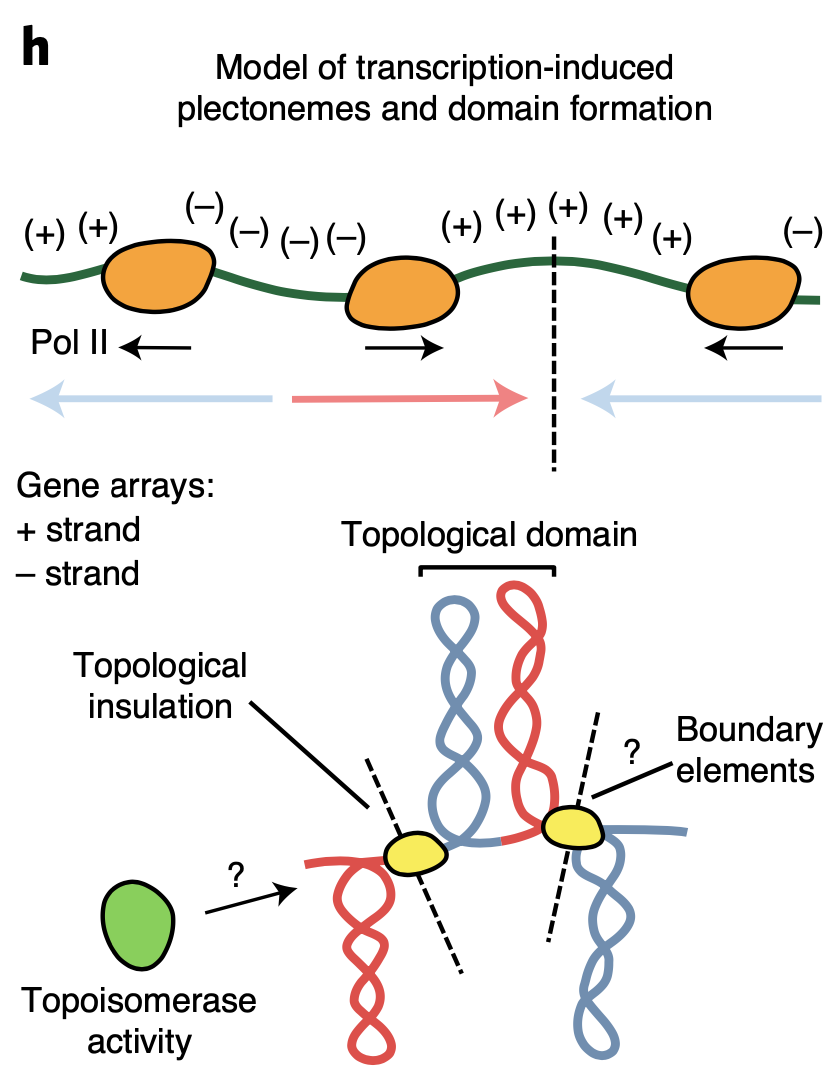
Georgi K. Marinov, Alexandro E. Trevino, Tingting Xiang, Anshul Kundaje, Arthur R. Grossman, William J. Greenleaf (2021) Nature Genetics.
Transcription-dependent domain-scale three-dimensional genome organization in the dinoflagellate Breviolum minutum
Dinoflagellate chromosomes represent a unique evolutionary experiment, as they exist in a permanently condensed, liquid crystalline state; are not packaged by histones; and contain genes organized into tandem gene arrays, with minimal transcriptional regulation. We analyze the three-dimensional genome of Breviolum minutum, and find large topological domains (dinoflagellate topologically associating domains, which we term ‘dinoTADs’) without chromatin loops, which are demarcated by convergent gene array boundaries. Transcriptional inhibition disrupts dinoTADs, implicating transcription-induced supercoiling as the primary topological force in dinoflagellates.
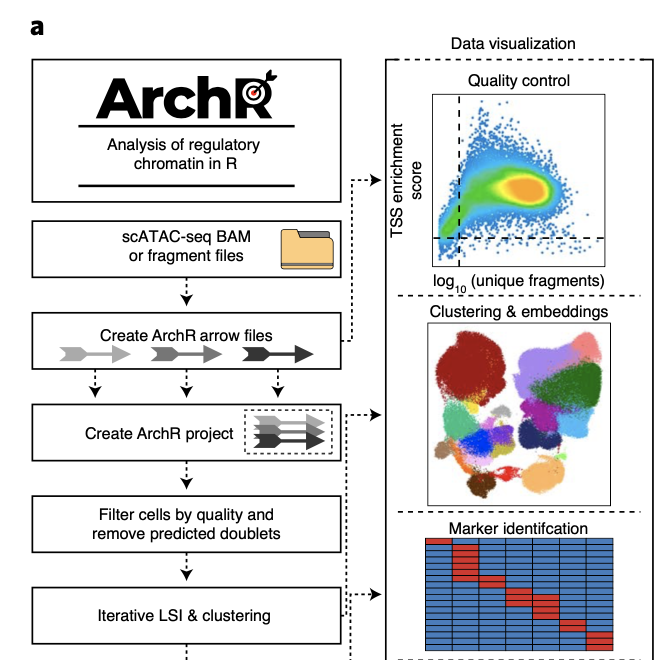
Jeffrey M. Granja, M. Ryan Corces, Sarah E. Pierce, S. Tansu Bagdatli, Hani Choudhry, Howard Y. Chang, William J. Greenleaf (2021) Nature Genetics.
ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis
The advent of single-cell chromatin accessibility profiling has accelerated the ability to map gene regulatory landscapes but has outpaced the development of scalable software to rapidly extract biological meaning from these data. Here we present a software suite for single-cell analysis of regulatory chromatin in R (ArchR; https://www.archrproject.com/) that enables fast and comprehensive analysis of single-cell chromatin accessibility data. ArchR provides an intuitive, user-focused interface for complex single-cell analyses, including doublet removal, single-cell clustering and cell type identification, unified peak set generation, cellular trajectory identification, DNA element-to-gene linkage, transcription factor footprinting, mRNA expres- sion level prediction from chromatin accessibility and multi-omic integration with single-cell RNA sequencing (scRNA-seq). Enabling the analysis of over 1.2 million single cells within 8 h on a standard Unix laptop, ArchR is a comprehensive software suite for end-to-end analysis of single-cell chromatin accessibility that will accelerate the understanding of gene regulation at the resolution of individual cells.
Evan A. Boyle, Winston R. Becker, Hua B. Bai, Janice S. Chen, Jennifer A. Doudna, William J. Greenleaf (2021) Science Advances
Quantification of Cas9 binding and cleavage across diverse guide sequences maps landscapes of target engagement
The RNA-guided nuclease Cas9 has unlocked powerful methods for perturbing both the genome through targeted DNA cleavage and the regulome through targeted DNA binding, but limited biochemical data have hampered efforts to quantitatively model sequence perturbation of target binding and cleavage across diverse guide sequences. We present scalable, sequencing-based platforms for high-throughput filter binding and cleavage and then perform 62,444 quantitative binding and cleavage assays on 35,047 on- and off-target DNA sequences across 90 Cas9 ribonucleoproteins (RNPs) loaded with distinct guide RNAs. We observe that binding and cleavage efficacy, as well as specificity, vary substantially across RNPs; canonically studied guides often have atypically high specificity; sequence context surrounding the target modulates Cas9 on-rate; and Cas9 RNPs may sequester targets in nonproductive states that contribute to “proofreading” capability. Lastly, we distill our findings into an interpretable biophysical model that predicts changes in binding and cleavage for diverse target sequence perturbations.
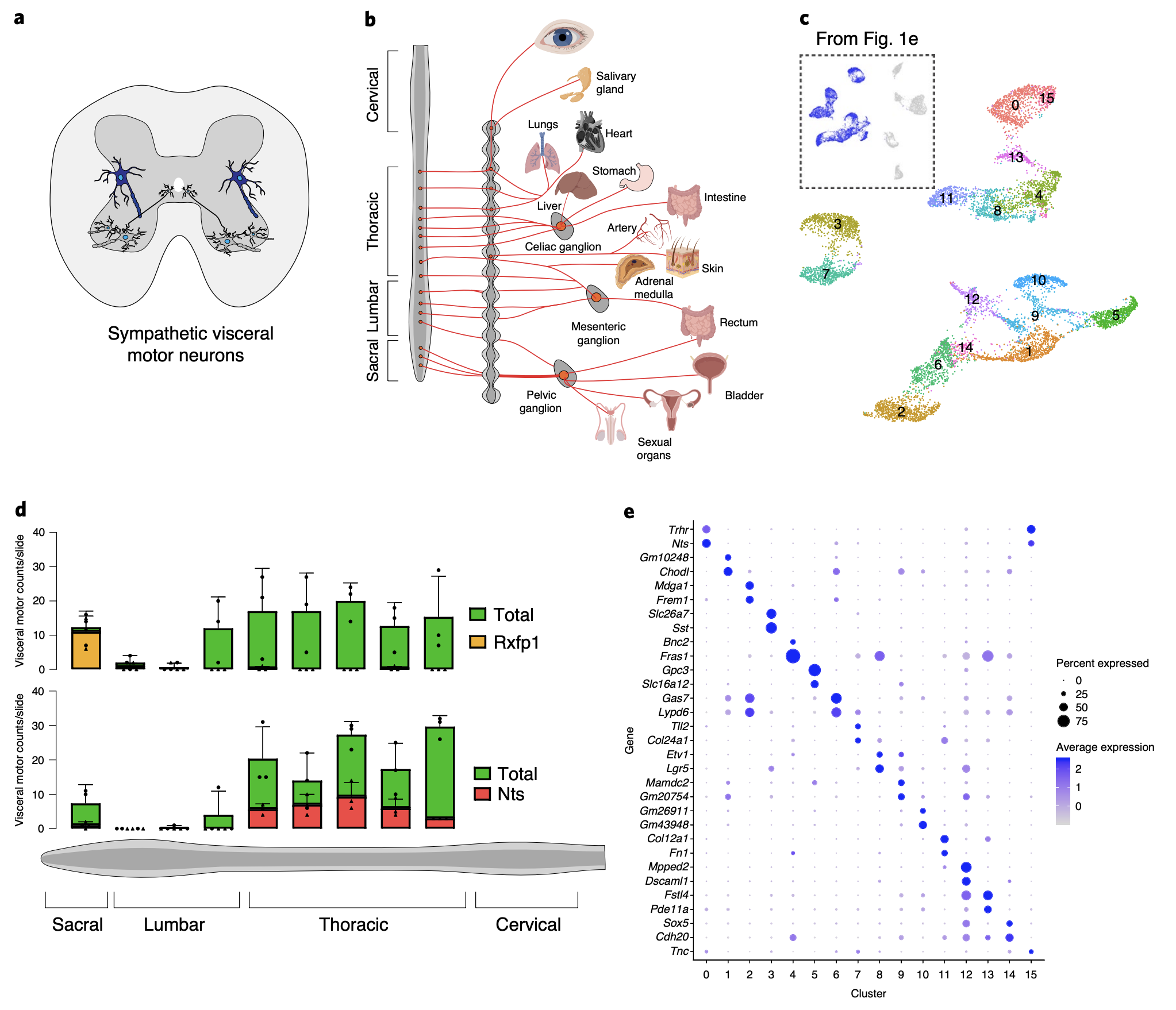
Jacob A. Blum, Sandy Klemm, Jennifer L. Shadrach, Kevin A. Guttenplan, Lisa Nakayama, Arwa Kathiria, Phuong T. Hoang, Olivia Gautier, Julia A. Kaltschmidt, William J. Greenleaf, Aaron D. Gitler (2021) Nature Neuroscience
Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons
The spinal cord is a fascinating structure that is responsible for coordinating movement in vertebrates. Spinal motor neurons control muscle activity by transmitting signals from the spinal cord to diverse peripheral targets. In this study, we profiled 43,890 single-nucleus transcriptomes from the adult mouse spinal cord using fluorescence-activated nuclei sorting to enrich for motor neuron nuclei. We identified 16 sympathetic motor neuron clusters, which are distinguishable by spatial localization and expression of neuromodulatory signaling genes. We found surprising skeletal motor neuron heterogeneity in the adult spinal cord, including transcriptional differences that correlate with electrophysiologically and spatially distinct motor pools. We also provide evidence for a novel transcriptional subpopulation of skeletal motor neuron (γ*). Collectively, these data provide a single-cell transcriptional atlas (http://spinalcordatlas.org) for investigating the organizing molecular logic of adult motor neuron diversity, as well as the cellular and molecular basis of motor neuron function in health and disease.
Maya Maor-Nof, Zohar Shipony, RodrigoLopez-Gonzalez, Lisa Nakayama, Yong-Jie Zhang, Julien Couthouis, Jacob A. Blum, Patricia A. Castruita, Gabriel R. Linares, Kai Ruan, Gokul Ramaswami, David J. Simon, Aviv Nof, Manuel Santana, Kyuho Han, Nasa Sinnott-Armstrong, Michael C. Bassik, Daniel H. Geschwind, Aaron D. Gitler. (2021) Cell
p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR)
The most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) is a GGGGCC repeat expansion in the C9orf72 gene. We developed a platform to interrogate the chromatin accessibility landscape and transcriptional program within neurons during degeneration. We provide evidence that neurons expressing the dipeptide repeat protein poly(proline-arginine), translated from the C9orf72 repeat expansion, activate a highly specific transcriptional program, exemplified by a single transcription factor, p53. Ablating p53 in mice completely rescued neurons from degeneration and markedly increased survival in a C9orf72 mouse model. p53 reduction also rescued axonal degeneration caused by poly(glycine-arginine), increased survival of C9orf72 ALS/FTD-patient-induced pluripotent stem cell (iPSC)-derived motor neurons, and mitigated neurodegeneration in a C9orf72 fly model. We show that p53 activates a downstream transcriptional program, including Puma, which drives neurodegeneration. These data demonstrate a neurodegenerative mechanism dynamically regulated through transcription-factor-binding events and provide a framework to apply chromatin accessibility and transcription program profiles to neurodegeneration.

Orit Rozenblatt-Rosen, Jay W. Shin, Jennifer E. Rood, Anna Hupalowska, Human Cell Atlas Standards and Technology Working Group, Aviv Regev, Holger Heyn (2021) “Building a high-quality Human Cell Atlas” Nature Biotechnology
Building a high-quality Human Cell Atlas
Building the Human Cell Atlas (HCA) requires consistent and agile experimental designs, standardized operating protocols (SOPs), benchmarks and quality control metrics that can adapt to a rapidly evolving technological landscape. Here, the HCA Standards and Technology Working Group outlines pertinent technical challenges and their approach to defining benchmarks and quality control measures to ensure high-quality data for building a comprehensive and accurate human cell atlas and help guide other atlas projects in health and disease.

Sarah E Pierce, Samuel H Kim, William J Greenleaf (2021) “Finding needles in a haystack: dissecting tumor heterogeneity with single-cell transcriptomic and chromatin accessibility profiling” Current Opinion in Genetics & Development
Finding needles in a haystack: dissecting tumor heterogeneity with single-cell transcriptomic and chromatin accessibility profiling
Tumor evolution often results in a wealth of heterogeneous cancer cell types within a single tumor — heterogeneity that can include epigenetic and gene expression changes that are impossible to identify from histological features alone. The invasion of cancer cells into nearby healthy tissue, accompanied by the infiltration of responding immune cells, results in an even more complex architecture of tumor and nontumor cells. However, bulk genomics-based methods can only assay the aggregate transcriptomic and epigenetic profiles across all of this rich cellular diversity. Such bulk averaging hides small subpopulations of tumor cells with unique phenotypes that might result in therapeutic resistance or metastatic progression. The advent of single-cell-based genomics assays for measuring transcription and chromatin accessibility – particularly scRNA-seq and scATAC-seq – has enabled the dissection of cell-types within tumors at a scale and resolution capable of unraveling the epigenetic and gene expression programs of rare and unique cellular subpopulations. This Review focuses on recent advances in scRNA-seq and scATAC-seq technologies and their application to cancer biology in the context of furthering our understanding of tumor heterogeneity.
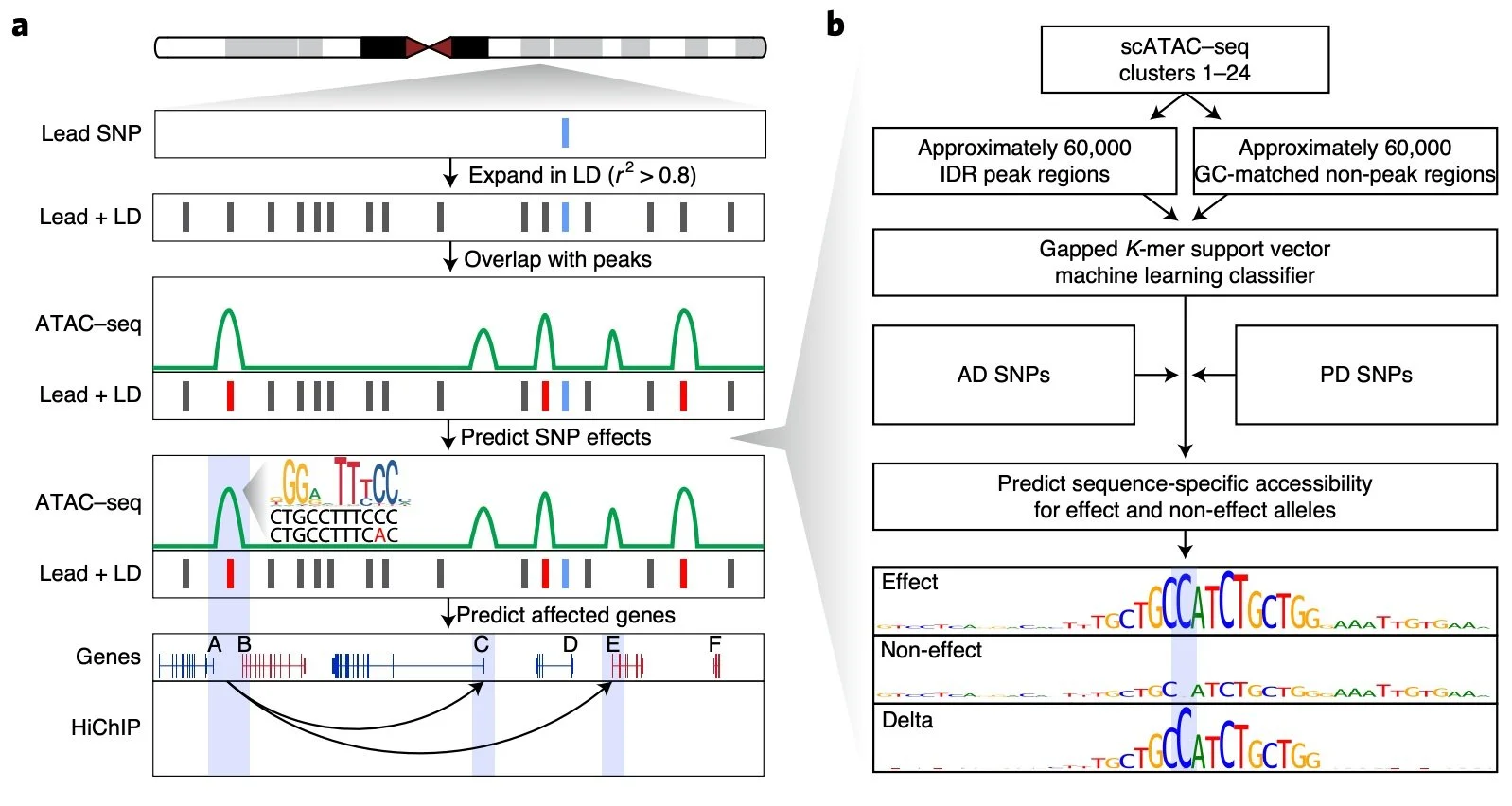
M. Ryan Corces, Anna Shcherbina, Soumya Kundu, Michael J. Gloudemans, Laure Frésard, Jeffrey M. Granja, Bryan H. Louie, Tiffany Eulalio, Shadi Shams, S. Tansu Bagdatli, Maxwell R. Mumbach, Boxiang Liu, Kathleen S. Montine, William J. Greenleaf, Anshul Kundaje, Stephen B. Montgomery, Howard Y. Chang, Thomas J. Montine (2020) “Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases” Nature Genetics
Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases
Genome-wide association studies of neurological diseases have identified thousands of variants associated with disease phenotypes. However, most of these variants do not alter coding sequences, making it difficult to assign their function. Here, we present a multi-omic epigenetic atlas of the adult human brain through profiling of single-cell chromatin accessibility landscapes and three-dimensional chromatin interactions of diverse adult brain regions across a cohort of cognitively healthy individuals. We developed a machine-learning classifier to integrate this multi-omic framework and predict dozens of functional SNPs for Alzheimer’s and Parkinson’s diseases, nominating target genes and cell types for previously orphaned loci from genome-wide association studies. Moreover, we dissected the complex inverted haplotype of the MAPT (encoding tau) Parkinson’s disease risk locus, identifying putative ectopic regulatory interactions in neurons that may mediate this disease association. This work expands understanding of inherited variation and provides a roadmap for the epigenomic dissection of causal regulatory variation in disease.
Johan O. L. Andreasson, Andrew Savinov, Steven M. Block, William J. Greenleaf (2020) “Comprehensive sequence-to-function mapping of cofactor-dependent RNA catalysis in the glmS ribozyme” Nature Communications
Comprehensive sequence-to-function mapping of cofactor-dependent RNA catalysis in the glmS ribozyme
Massively parallel, quantitative measurements of biomolecular activity across sequence space can greatly expand our understanding of RNA sequence-function relationships. We report the development of an RNA-array assay to perform such measurements and its application to a model RNA: the core glmS ribozyme riboswitch, which performs a ligand- dependent self-cleavage reaction. We measure the cleavage rates for all possible single and double mutants of this ribozyme across a series of ligand concentrations, determining kcat and KM values for active variants. These systematic measurements suggest that evolutionary conservation in the consensus sequence is driven by maintenance of the cleavage rate. Analysis of double-mutant rates and associated mutational interactions produces a structural and functional mapping of the ribozyme sequence, revealing the catalytic consequences of specific tertiary interactions, and allowing us to infer structural rearrangements that permit certain sequence variants to maintain activity.
Zohar Shipony, Georgi K. Marinov, Matthew P. Swaffer, Nicholas A. Sinnott-Armstrong, Jan M. Skotheim, Anshul Kundaje, William J. Greenleaf (2020) “Long-range single-molecule mapping of chromatin accessibility in eukaryotes” Nature Methods
Long-range single-molecule mapping of chromatin accessibility in eukaryotes
Mapping open chromatin regions has emerged as a widely used tool for identifying active regulatory elements in eukaryotes. However, existing approaches, limited by reliance on DNA fragmentation and short-read sequencing, cannot provide informa- tion about large-scale chromatin states or reveal coordination between the states of distal regulatory elements. We have developed a method for profiling the accessibility of individual chromatin fibers, a single-molecule long-read accessible chromatin mapping sequencing assay (SMAC-seq), enabling the simultaneous, high-resolution, single-molecule assessment of chroma- tin states at multikilobase length scales. Our strategy is based on combining the preferential methylation of open chromatin regions by DNA methyltransferases with low sequence specificity, in this case EcoGII, an N6-methyladenosine (m6A) methyltransferase, and the ability of nanopore sequencing to directly read DNA modifications. We demonstrate that aggregate SMAC-seq signals match bulk-level accessibility measurements, observe single-molecule nucleosome and transcription factor protection footprints, and quantify the correlation between chromatin states of distal genomic elements.

Alexandro E. Trevino, Nasa Sinnott-Armstrong, Jimena Andersen, Se-Jin Yoon, Nina Huber, Jonathan K. Pritchard, Howard Y. Chang, William J. Greenleaf, Sergiu P. Pașca (2020) “Chromatin accessibility dynamics in a model of human forebrain development” Science
Chromatin accessibility dynamics in a model of human forebrain development
Forebrain development is characterized by highly synchronized cellular processes, which, if perturbed, can cause disease. To chart the regulatory activity underlying these events, we generated a map of accessible chromatin in human three-dimensional forebrain organoids. To capture corticogenesis, we sampled glial and neuronal lineages from dorsal or ventral forebrain organoids over 20 months in vitro. Active chromatin regions identified in human primary brain tissue were observed in organoids at different developmental stages. We used this resource to map genetic risk for disease and to explore evolutionary conservation. Moreover, we integrated chromatin accessibility with transcriptomics to identify putative enhancer-gene linkages and transcription factors that regulate human corticogenesis. Overall, this platform brings insights into gene-regulatory dynamics at previously inaccessible stages of human forebrain development, including signatures of neuropsychiatric disorders.
Jeffrey M. Granja, Sandy Klemm, Lisa M. McGinnis, Arwa S. Kathiria, Anja Mezger, M. Ryan Corces, Benjamin Parks, Eric Gars, Michaela Liedtke, Grace X. Y. Zheng, Howard Y. Chang, Ravindra Majeti, and William J. Greenleaf (2019) “Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia” Nature Biotechnology
Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia
Identifying the causes of human diseases requires deconvolution of abnormal molecular phenotypes spanning DNA accessibility, gene expression and protein abundance. We present a single-cell framework that integrates highly multiplexed protein quantification, transcriptome profiling and analysis of chromatin accessibility. Using this approach, we establish a normal epigenetic baseline for healthy blood development, which we then use to deconvolve aberrant molecular features within blood from patients with mixed-phenotype acute leukemia. Despite widespread epigenetic heterogeneity within the patient cohort, we observe common malignant signatures across patients as well as patient-specific regulatory features that are shared across phenotypic compartments of individual patients. Integrative analysis of transcriptomic and chromatin-accessibility maps identified 91,601 putative peak-to-gene linkages and transcription factors that regulate leukemia specific genes, such as RUNX1-linked regulatory elements proximal to the marker gene CD69. These results demonstrate how integrative, multiomic analysis of single cells within the framework of normal development can reveal both distinct and shared molecular mechanisms of disease from patient samples.
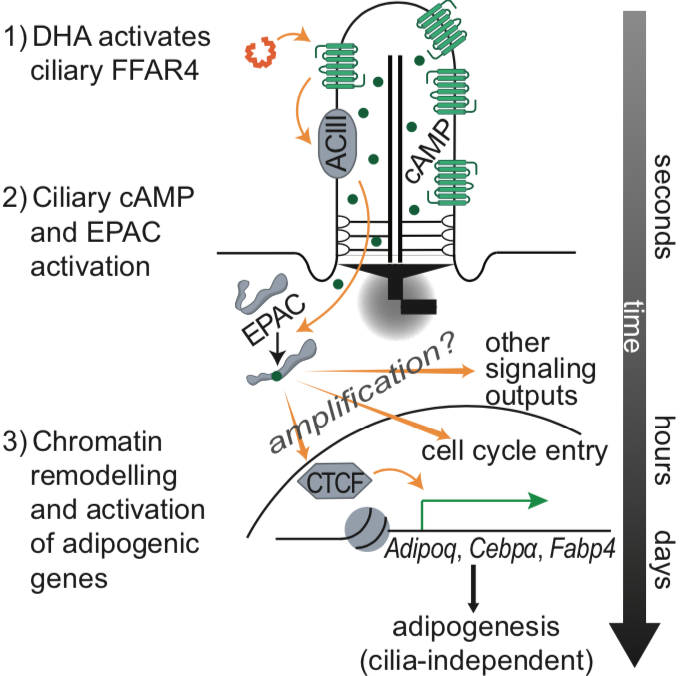
Keren I Hilgendorf, Carl T Johnson, Anja Mezger, Selena L Rice, Alessandra M Norris, Janos Demeter, William J Greenleaf, Jeremy F Reiter, Daniel Kopinke, Peter K Jackson. (2019) “Omega-3 Fatty Acids Activate Ciliary FFAR4 to Control Adipogenesis” Cell
Omega-3 Fatty Acids Activate Ciliary FFAR4 to Control Adipogenesis
Adult mesenchymal stem cells, including preadipocytes, possess a cellular sensory organelle called the primary cilium. Ciliated preadipocytes abundantly populate perivascular compartments in fat and are activated by a high-fat diet. Here, we sought to understand whether preadipocytes use their cilia to sense and respond to external cues to remodel white adipose tissue. Abolishing preadipocyte cilia in mice severely impairs white adipose tissue expansion. We discover that TULP3-dependent ciliary localization of the omega-3 fatty acid receptor FFAR4/GPR120 promotes adipogenesis. FFAR4 agonists and ω-3 fatty acids, but not saturated fatty acids, trigger mitosis and adipogenesis by rapidly activating cAMP production inside cilia. Ciliary cAMP activates EPAC signaling, CTCF-dependent chromatin remodeling, and transcriptional activation of PPARγ and CEBPα to initiate adipogenesis. We propose that dietary ω-3 fatty acids selectively drive expansion of adipocyte numbers to produce new fat cells and store saturated fatty acids, enabling homeostasis of healthy fat tissue.
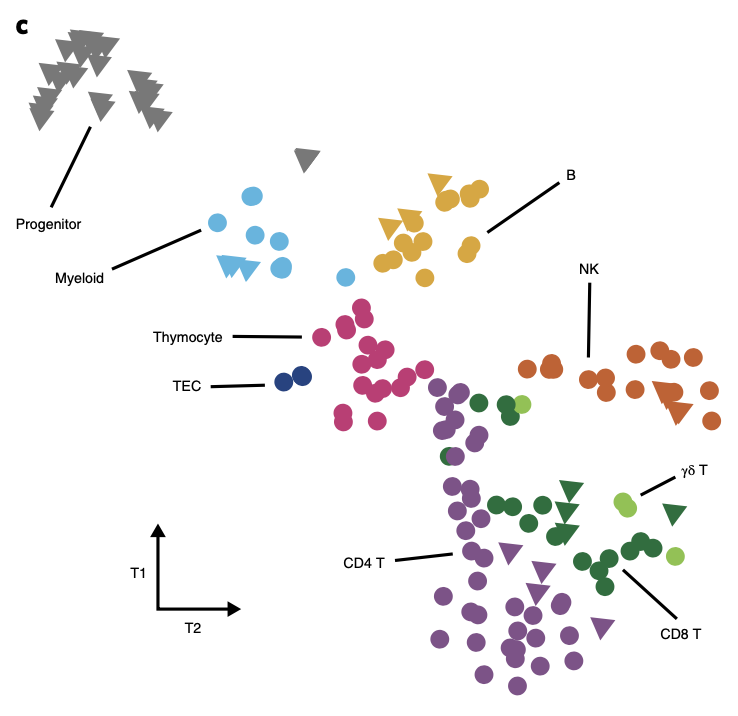
Diego Calderon, Michelle L. T. Nguyen, Anja Mezger, Arwa Kathiria, Fabian Müller, Vinh Nguyen, Ninnia Lescano, Beijing Wu, John Trombetta, Jessica V. Ribado, David A. Knowles, Ziyue Gao, Franziska Blaeschke, Audrey V. Parent, Trevor D. Burt, Mark S. Anderson, Lindsey A. Criswell*, William J. Greenleaf*, Alexander Marson*, and Jonathan K. Pritchard* (2020) “Landscape of stimulation-responsive chromatin across diverse human immune cells” Nature Genetics
Landscape of stimulation-responsive chromatin across diverse human immune cells
A hallmark of the immune system is the interplay among specialized cell types transitioning between resting and stimulated states. The gene regulatory landscape of this dynamic system has not been fully characterized in human cells. Here we collected assay for transposase-accessible chromatin using sequencing (ATAC-seq) and RNA sequencing data under resting and stimulated conditions for up to 32 immune cell populations. Stimulation caused widespread chromatin remodeling, including response elements shared between stimulated B and T cells. Furthermore, several autoimmune traits showed significant heritability in stimulation- responsive elements from distinct cell types, highlighting the importance of these cell states in autoimmunity. Allele-specific read mapping identified variants that alter chromatin accessibility in particular conditions, allowing us to observe evidence of function for a candidate causal variant that is undetected by existing large-scale studies in resting cells. Our results provide a resource of chromatin dynamics and highlight the need to characterize the effects of genetic variation in stimulated cells.
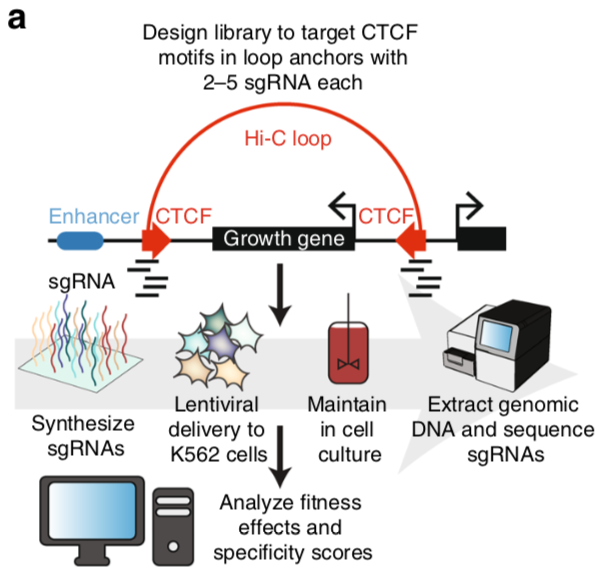
Josh Tycko, Michael Wainberg, Georgi K. Marinov, Oana Ursu, Gaelen T. Hess, Braeden K. Ego, Aradhana, Amy Li, Alisa Truong, Alexandro E. Trevino, Kaitlyn Spees, David Yao, Irene M. Kaplow, Peyton G. Greenside, David W. Morgens, Douglas H. Phanstiel, Michael P. Snyder, William J. Greenleaf, Anshul Kundaje, Michael C. Bassik (2019) “Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements” Nature Communications
Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements
Pooled CRISPR-Cas9 screens are a powerful method for functionally characterizing regulatory elements in the non-coding genome, but off-target effects in these experiments have not been systematically evaluated. Here, we investigate Cas9, dCas9, and CRISPRi/a off-target activity in screens for essential regulatory elements. The sgRNAs with the largest effects in genome-scale screens for essential CTCF loop anchors in K562 cells were not single guide RNAs (sgRNAs) that disrupted gene expression near the on-target CTCF anchor. Rather, these sgRNAs had high off-target activity that, while only weakly correlated with absolute off- target site number, could be predicted by the recently developed GuideScan specificity score. Screens conducted in parallel with CRISPRi/a, which do not induce double-stranded DNA breaks, revealed that a distinct set of off-targets also cause strong confounding fitness effects with these epigenome-editing tools. Promisingly, filtering of CRISPRi libraries using Guide- Scan specificity scores removed these confounded sgRNAs and enabled identification of essential regulatory elements.
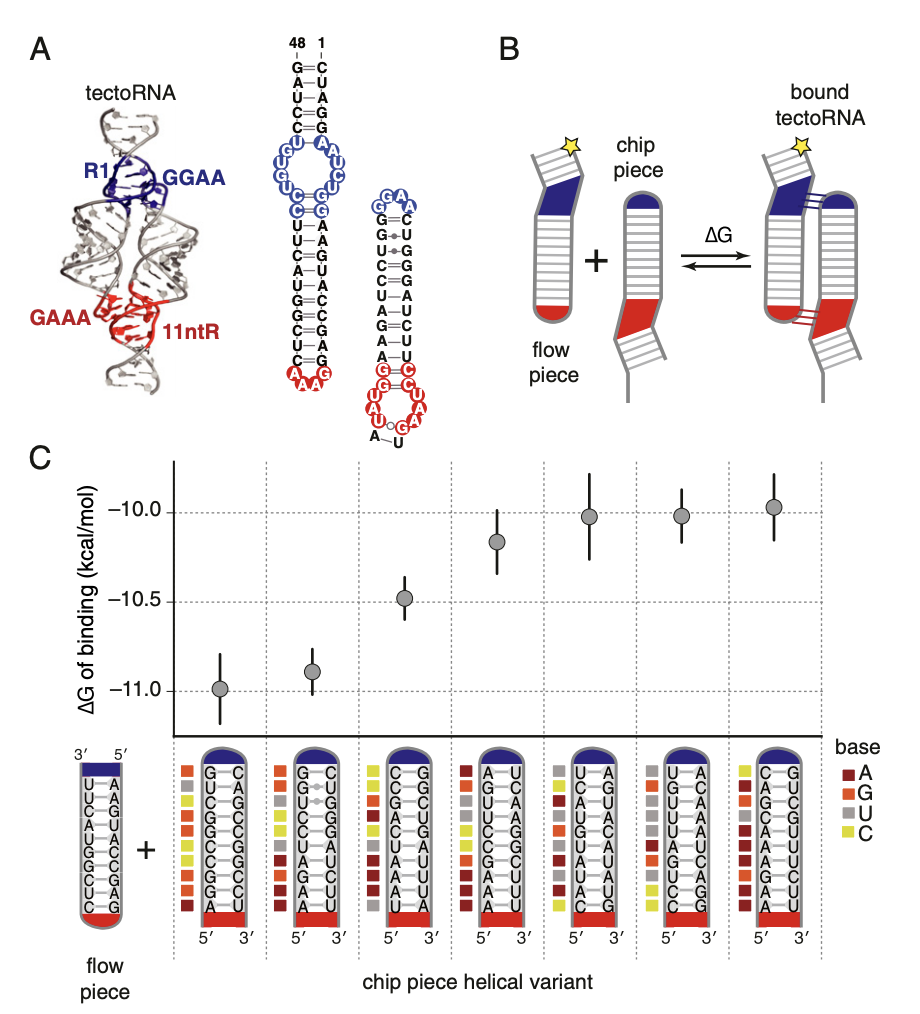
Joseph D. Yesselman, Sarah K. Denny, Namita Bisaria, Daniel Herschlag, William J. Greenleaf, and Rhiju Das (2019) “ Sequence-dependent RNA helix conformational preferences predictably impact tertiary structure formation ” PNAS
Sequence-dependent RNA helix conformational preferences predictably impact tertiary structure formation
Structured RNAs fold into complex tertiary structures to perform critical roles in a multitude of biological functions. Over half the nucleotides in structured RNAs form simple Watson–Crick (WC) double helices, which can then assemble through non-WC interactions into elaborate tertiary structures. Here, we report the serendipitous discovery that sequence changes in WC base pairs impact the energetics of RNA tertiary folding. These observations led to a computational model for helix conformational fluctuations that then blindly predicted the results of thousands of high-throughput experiments with surprisingly high accuracy. Our study reveals that sequence-specific helix structure preferences are needed for understanding RNA folding quantitatively and outlines a route for dissecting the impact of helix conformational fluctuations across general RNA biophysical events.
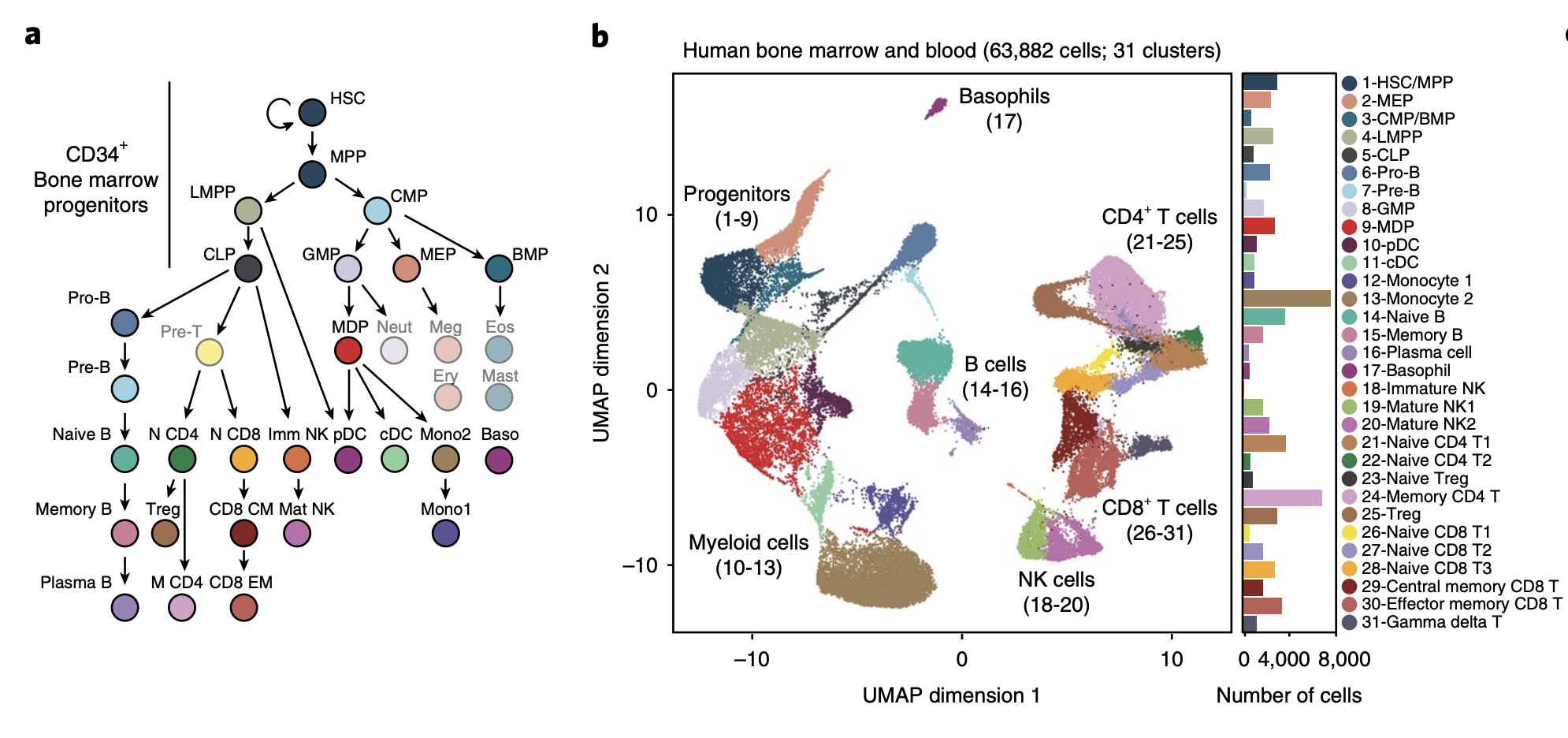
Ansuman T. Satpathy, Jeffrey M. Granja, Kathryn E. Yost, Yanyan Qi, Francesca Meschi, Geoffrey P. McDermott, Brett N. Olsen, Maxwell R. Mumbach, Sarah E. Pierce, M. Ryan Corces, Preyas Shah, Jason C. Bell, Darisha Jhutty, Corey M. Nemec, Jean Wang, Li Wang, Yifeng Yin, Paul G. Giresi, Anne Lynn S. Chang, Grace X. Y. Zheng, William J. Greenleaf & Howard Y. Chang (2019) “ Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion ” Nature Biotechnology
Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion
Understanding complex tissues requires single-cell deconstruction of gene regulation with precision and scale. Here, we assess the performance of a massively parallel droplet-based method for mapping transposase-accessible chromatin in single cells using sequencing (scATAC-seq). We apply scATAC-seq to obtain chromatin profiles of more than 200,000 single cells in human blood and basal cell carcinoma. In blood, application of scATAC-seq enables marker-free identification of cell type-specific cis- and trans-regulatory elements, mapping of disease-associated enhancer activity and reconstruction of trajectories of cellular differentiation. In basal cell carcinoma, application of scATAC-seq reveals regulatory networks in malignant, stromal and immune cells in the tumor microenvironment. Analysis of scATAC-seq profiles from serial tumor biopsies before and after programmed cell death protein 1 blockade identifies chromatin regulators of therapy-responsive T cell subsets and reveals a shared regulatory program that governs intratumoral CD8+ T cell exhaustion and CD4+ T follicular helper cell development. Weanticipate that scATAC-seq will enable the unbiased discovery of gene regulatory factors across diverse biological systems.
Winston R. Becker, Benjamin Ober-Reynolds, Karina Jouravleva, Samson M. Jolly, Phillip D. Zamore, William J. Greenleaf, (2019) “ High-Throughput Analysis Reveals Rules for Target RNA Binding and Cleavage by AGO2 ” Molecular Cell
High-Throughput Analysis Reveals Rules for Target RNA Binding and Cleavage by AGO2
Argonaute proteins loaded with microRNAs (miRNAs) or small interfering RNAs (siRNAs) form the RNA-induced silencing complex (RISC), which represses target RNA expression. Predicting the biological targets, specificity, and efficiency of both miRNAs and siRNAs has been hamstrung by an incomplete understanding of the sequence determinants of RISC binding and cleavage. We applied high-throughput methods to measure the association kinetics, equilibrium binding energies, and single-turnover cleavage rates of mouse AGO2 RISC. We find that RISC readily tolerates insertions of up to 7 nt in its target opposite the central region of the guide. Our data uncover specific guide:target mismatches that enhance the rate of target cleavage, suggesting novel siRNA design strategies. Using these data, we derive quantitative models for RISC binding and target cleavage and show that our in vitro measurements and models predict knockdown in an engineered cellular system.
Rohit R. Jadhav, Se Jin Im, Bin Hu, Masao Hashimoto, Peng Li, Jian-Xin Lin, Warren J. Leonard, William J. Greenleaf, Rafi Ahmed, Jorg J. Goronzy. (2019) “Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade” PNAS
Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade
We have recently defined a novel population of PD-1 (programmed cell death 1)+ TCF1 (T cell factor 1)+ virus-specific CD8 T cells that function as resource cells during chronic LCMV infection and provide the proliferative burst seen after PD-1 blockade. Such CD8 T cells have been found in other chronic infections and also in cancer in mice and humans. These CD8 T cells exhibit stem-like properties undergoing self-renewal and also differentiating into the terminally exhausted CD8 T cells. Here we compared the epigenetic signature of stem-like CD8 T cells with exhausted CD8 T cells. ATAC-seq analysis showed that stem-like CD8 T cells had a unique signature implicating activity of HMG (TCF) and RHD (NF-κB) transcription factor family members in contrast to higher accessibility to ETS and RUNX motifs in exhausted CD8 T cells. In addition, regulatory regions of the transcription factors Tcf7 and Id3 were more accessible in stem-like cells whereas Prdm1 and Id2 were more accessible in exhausted CD8 T cells. We also compared the epigenetic signatures of the 2 CD8 T cell subsets from chronically infected mice with effector and memory CD8 T cells generated after an acute LCMV infection. Both CD8 T cell subsets generated during chronic infection were strikingly different from CD8 T cell subsets from acute infection. Interestingly, the stem-like CD8 T cell subset from chronic infection, despite sharing key functional properties with memory CD8 T cells, had a very distinct epigenetic program. These results show that the chronic stem-like CD8 T cell program represents a specific adaptation of the T cell response to persistent antigenic stimulation.
Maxwell R. Mumbach, Jeffrey M. Granja, Ryan A. Flynn, Caitlin M. Roake, Ansuman T. Satpathy, Adam J. Rubin, Yanyan Qi, Zhaozhao Jiang, Shadi Shams, Bryan H. Louie, Jimmy K. Guo, David G. Gennert, M. Ryan Corces, Paul A. Khavari, Maninjay K. Atianand, Steven E. Artandi, Katherine A. Fitzgerald, William J. Greenleaf & Howard Y. Chang (2019) “ HiChIRP reveals RNA-associated chromosome conformation ” Nature Methods
HiChIRP reveals RNA-associated chromosome conformation
Modular domains of long non-coding RNAs can serve as scaffolds to bring distant regions of the linear genome into spatial proximity. Here, we present HiChIRP, a method leveraging bio-orthogonal chemistry and optimized chromosome conformation capture conditions, which enables interrogation of chromatin architecture focused around a specific RNA of interest down to approximately ten copies per cell. HiChIRP of three nuclear RNAs reveals insights into promoter interactions (7SK), telomere biology (telomerase RNA component) and inflammatory gene regulation (lincRNA-EPS).
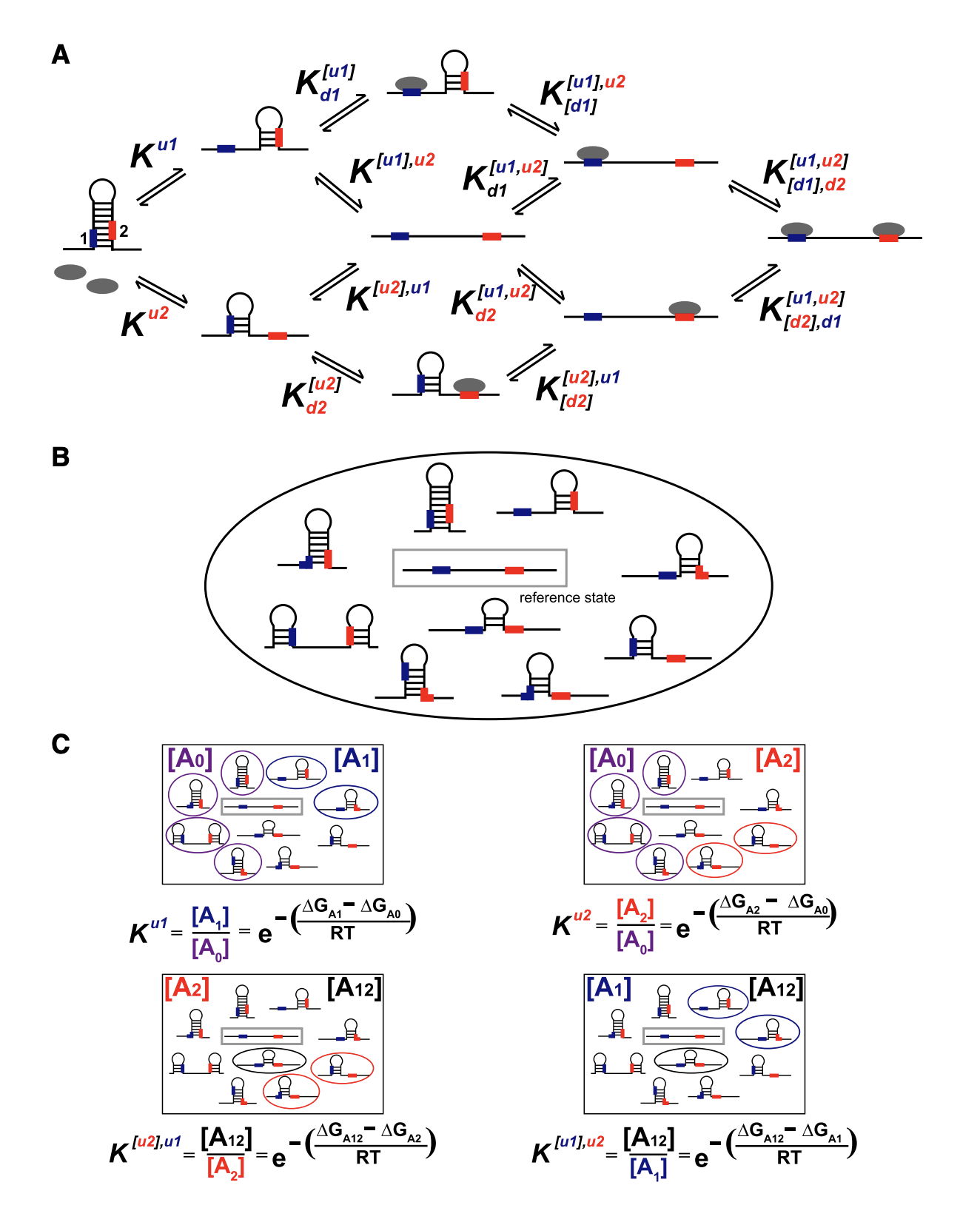
Winston R. Becker, Inga Jarmoskaite, Pavanapuresan P. Vaidyanathan, William J. Greenleaf and Daniel Herschlag (2019) “ Demonstration of protein cooperativity mediated by RNA structure using the human protein PUM2 ” RNA
Demonstration of protein cooperativity mediated by RNA structure using the human protein PUM2
Posttranslational gene regulation requires a complex network of RNA–protein interactions. Cooperativity, which tunes response sensitivities, originates from protein–protein interactions in many systems. For RNA-binding proteins, cooperativity can also be mediated through RNA structure. RNA structural cooperativity (RSC) arises when binding of one protein induces a redistribution of RNA conformational states that enhance access (positive cooperativity) or block access (negative cooperativity) to additional binding sites. As RSC does not require direct protein–protein interactions, it allows cooperativity to be tuned for individual RNAs, via alterations in sequence that alter structural stability. Given the potential importance of this mechanism of control and our desire to quantitatively dissect features that underlie physiological regulation, we developed a statistical mechanical framework for RSC and tested this model by performing equilibrium binding measurements of the human PUF family protein PUM2. Using 68 RNAs that contain two to five PUM2-binding sites and RNA structures of varying stabilities, we observed a range of structure-dependent cooperative behaviors. To test our ability to account for this cooperativity with known physical constants, we used PUM2 affinity and nearest-neighbor RNA secondary structure predictions. Our model gave qualitative agreement for our disparate set of 68 RNAs across two temperatures, but quantitative deviations arise from overestimation of RNA structural stability. Our results demonstrate cooperativity mediated by RNA structure and underscore the power of quantitative stepwise experimental evaluation of mechanisms and computational tools.
Inga Jarmoskaite, Sarah K. Denny, Pavanapuresan P. Vaidyanathan, Winston R. Becker, Johan O.L. Andreasson, Curtis J. Layton, Kalli Kappe, Varun Shivashankar, Raashi Sreenivasan, Rhiju Das, William J.Greenleaf, Daniel Herschlag (2019) “ A Quantitative and Predictive Model for RNA Binding by Human Pumilio Proteins ” Molecular Cell
A Quantitative and Predictive Model for RNA Binding by Human Pumilio Proteins
High-throughput methodologies have enabled routine generation of RNA target sets and sequence motifs for RNA-binding proteins (RBPs). Nevertheless, quantitative approaches are needed to capture the landscape of RNA-RBP interactions responsible for cellular regulation. We have used the RNA-MaP platform to directly measure equilibrium binding for thousands of designed RNAs and to construct a predictive model for RNA recognition by the human Pumilio proteins PUM1 and PUM2. Despite prior findings of linear sequence motifs, our measurements revealed widespread residue flipping and instances of positional coupling. Application of our thermodynamic model to published in vivo crosslinking data reveals quantitative agreement between predicted affinities and in vivo occupancies. Our analyses suggest a thermodynamically driven, continuous Pumilio-binding landscape that is negligibly affected by RNA structure or kinetic factors, such as displacement by ribosomes. This work provides a quantitative foundation for dissecting the cellular behavior of RBPs and cellular features that impact their occupancies.
Curtis J.Layton, Peter L. McMahon, William J.Greenleaf (2019) “ Large-Scale, Quantitative Protein Assays on a High-Throughput DNA Sequencing Chip ” Molecular Cell
Large-Scale, Quantitative Protein Assays on a HighThroughput DNA Sequencing Chip
High-throughput DNA sequencing techniques have enabled diverse approaches for linking DNA sequence to biochemical function. In contrast, assays of protein function have substantial limitations in terms of throughput, automation, and widespread availability. We have adapted an Illumina high-throughput sequencing chip to display an immense diversity of ribosomally translated proteins and peptides and then carried out fluorescence-based functional assays directly on this flow cell, demonstrating that a single, widely available high-throughput platform can perform both sequencing-by-synthesis and protein assays. We quantified the binding of the M2 anti-FLAG antibody to a library of 1.3 × 104 variant FLAG peptides, exploring non-additive effects of combinations of mutations and discovering a “superFLAG” epitope variant. We also measured the enzymatic activity of 1.56 × 105 molecular variants of full-length human O6-alkylguanine-DNA alkyltransferase (SNAP-tag). This comprehensive corpus of catalytic rates revealed amino acid interaction networks and cooperativity, linked positive cooperativity to structural proximity, and revealed ubiquitous positively cooperative interactions with histidine residues.
Sandy L. Klemm, Zohar Shipony & William J. Greenleaf (2019) “ Chromatin accessibility and the regulatory epigenome ” Nature Reviews Genetics
Chromatin accessibility and the regulatory epigenome
Physical access to DNA is a highly dynamic property of chromatin that plays an essential role in establishing and maintaining cellular identity. The organization of accessible chromatin across the genome reflects a network of permissible physical interactions through which enhancers, promoters, insulators and chromatin-binding factors cooperatively regulate gene expression. This landscape of accessibility changes dynamically in response to both external stimuli and developmental cues, and emerging evidence suggests that homeostatic maintenance of accessibility is itself dynamically regulated through a competitive interplay between chromatin-binding factors and nucleosomes. In this Review, we examine how the accessible genome is measured and explore the role of transcription factors in initiating accessibility remodelling; our goal is to illustrate how chromatin accessibility defines regulatory elements within the genome and how these epigenetic features are dynamically established to control gene expression.
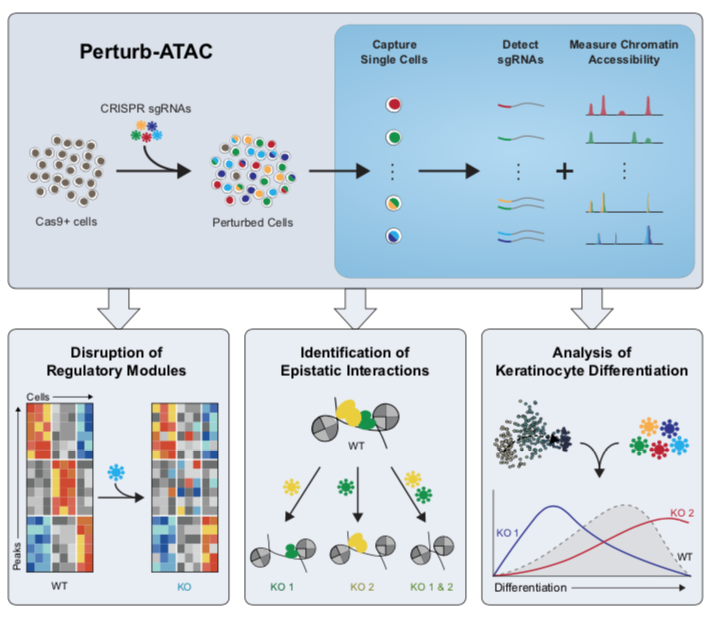
Adam J. Rubin, Kevin R. Parker, Ansuman T. Satpathy, Yanyan Qi, Beijing Wu, Alvin J. Ong, Maxwell R. Mumbach, Andrew L. Ji, Daniel S. Kim, Seung Woo Cho, Brian J. Zarnegar, William J. Greenleaf, Howard Y. Chang, Paul A. Khavari. (2019) “Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks” Cell
Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks
Here, we present Perturb-ATAC, a method that combines multiplexed CRISPR interference or knockout with genome-wide chromatin accessibility profiling in single cells based on the simultaneous detection of CRISPR guide RNAs and open chromatin sites by assay of transposase-accessible chromatin with sequencing (ATAC-seq). We applied Perturb-ATAC to transcription factors (TFs), chromatin-modifying factors, and noncoding RNAs (ncRNAs) in ∼4,300 single cells, encompassing more than 63 genotype-phenotype relationships. Perturb-ATAC in human B lymphocytes uncovered regulators of chromatin accessibility, TF occupancy, and nucleosome positioning and identified a hierarchy of TFs that govern B cell state, variation, and disease-associated cis-regulatory elements. Perturb-ATAC in primary human epidermal cells revealed three sequential modules of cis-elements that specify keratinocyte fate. Combinatorial deletion of all pairs of these TFs uncovered their epistatic relationships and highlighted genomic co-localization as a basis for synergistic interactions. Thus, Perturb-ATAC is a powerful strategy to dissect gene regulatory networks in development and disease.
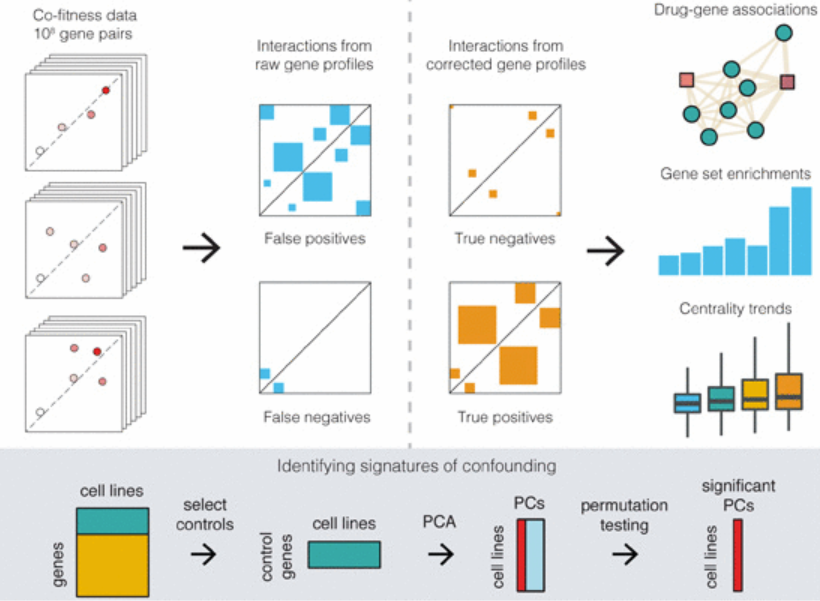
Evan A Boyle , Jonathan K Pritchard & William J Greenleaf (2018) “ High‐resolution mapping of cancer cell networks using co‐functional interactions ” Molecular Systems Biology
High‐resolution mapping of cancer cell networks using co‐functional interactions
Powerful new technologies for perturbing genetic elements have recently expanded the study of genetic interactions in model systems ranging from yeast to human cell lines. However, technical artifacts can confound signal across genetic screens and limit the immense potential of parallel screening approaches. To address this problem, we devised a novel PCA‐based method for correcting genome‐wide screening data, bolstering the sensitivity and specificity of detection for genetic interactions. Applying this strategy to a set of 436 whole genome CRISPR screens, we report more than 1.5 million pairs of correlated “co‐functional” genes that provide finer‐scale information about cell compartments, biological pathways, and protein complexes than traditional gene sets. Lastly, we employed a gene community detection approach to implicate core genes for cancer growth and compress signal from functionally related genes in the same community into a single score. This work establishes new algorithms for probing cancer cell networks and motivates the acquisition of further CRISPR screen data across diverse genotypes and cell types to further resolve complex cellular processes.
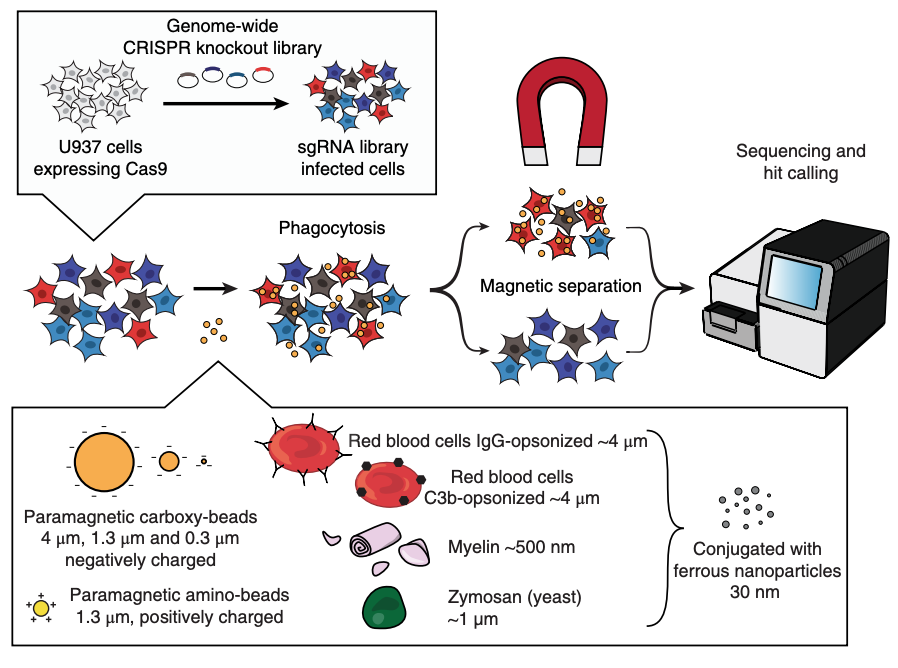
Michael S. Haney, Christopher J. Bohlen, David W. Morgens, James A. Ousey, Amira A. Barkal, C. Kimberly Tsui, Braeden K. Ego, Roni Levin, Roarke A. Kamber, Hannah Collins, Andrew Tucker, Amy Li, Daan Vorselen, Lorenzo Labitigan, Emily Crane, Evan Boyle, Lihua Jiang, Joanne Chan, Esther Rincón, William J. Greenleaf, Billy Li, Michael P. Snyder, Irving L. Weissman, Julie A. Theriot, Sean R. Collins, Ben A. Barres, Michael C. Bassik. (2018) “Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens” Nature Genetics
Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens
Phagocytosis is required for a broad range of physiological functions, from pathogen defense to tissue homeostasis, but the mechanisms required for phagocytosis of diverse substrates remain incompletely understood. Here, we developed a rapid magnet-based phenotypic screening strategy, and performed eight genome-wide CRISPR screens in human cells to identify genes regulating phagocytosis of distinct substrates. After validating select hits in focused miniscreens, orthogonal assays and primary human macrophages, we show that (1) the previously uncharacterized gene NHLRC2 is a central player in phagocytosis, regulating RhoA-Rac1 signaling cascades that control actin polymerization and filopodia formation, (2) very-long-chain fatty acids are essential for efficient phagocytosis of certain substrates and (3) the previously uncharacterized Alzheimer’s disease–associated gene TM2D3 can preferentially influence uptake of amyloid-β aggregates. These findings illuminate new regulators and core principles of phagocytosis, and more generally establish an efficient method for unbiased identification of cellular uptake mechanisms across diverse physiological and pathological contexts.
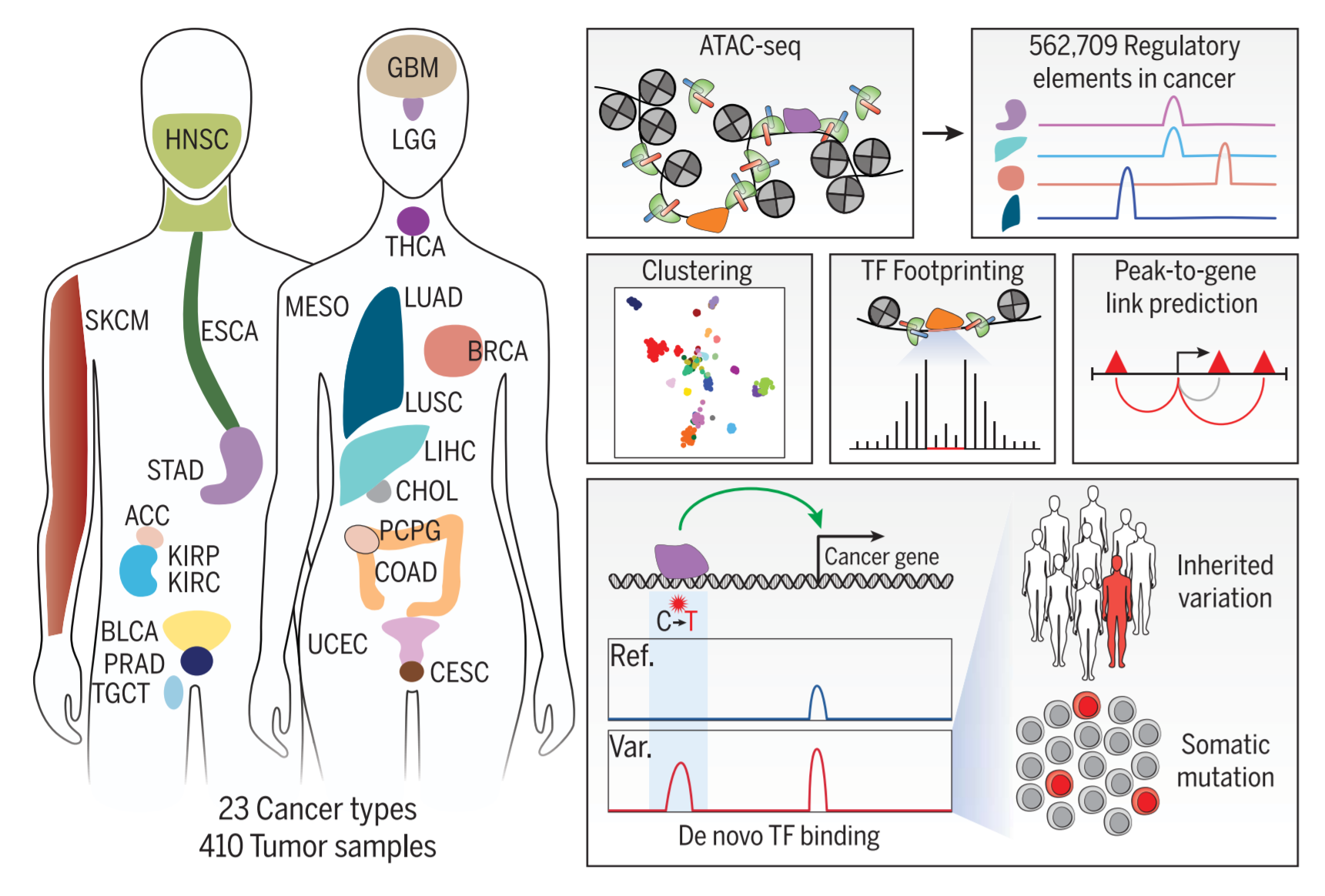
M. Ryan Corces, Jeffrey M. Granja, Shadi Shams, Bryan H. Louie, Jose A. Seoane, Wanding Zhou, Tiago C. Silva, Clarice Groeneveld, Christopher K. Wong, Seung Woo Cho, Ansuman T. Satpathy, Maxwell R. Mumbach, Katherine A. Hoadley, A. Gordon Robertson, Nathan C. Sheffield, Ina Felau, Mauro A. A. Castro, Benjamin P. Berman, Louis M. Staudt, Jean C. Zenklusen, Peter W. Laird, Christina Curtis, The Cancer Genome Atlas Analysis Network, William J. Greenleaf, Howard Y. Chang (2018) “ The chromatin accessibility landscape of primary human cancers ” Science
The chromatin accessibility landscape of primary human cancers
The Cancer Genome Atlas (TCGA) provides a high-quality resource of molecular data on a large variety of human cancers. Corces et al. used a recently modified assay to profile chromatin accessibility to determine the accessible chromatin landscape in 410 TCGA samples from 23 cancer types (see the Perspective by Taipale). When the data were integrated with other omics data available for the same tumor samples, inherited risk loci for cancer predisposition were revealed, transcription factors and enhancers driving molecular subtypes of cancer with patient survival differences were identified, and noncoding mutations associated with clinical prognosis were discovered.
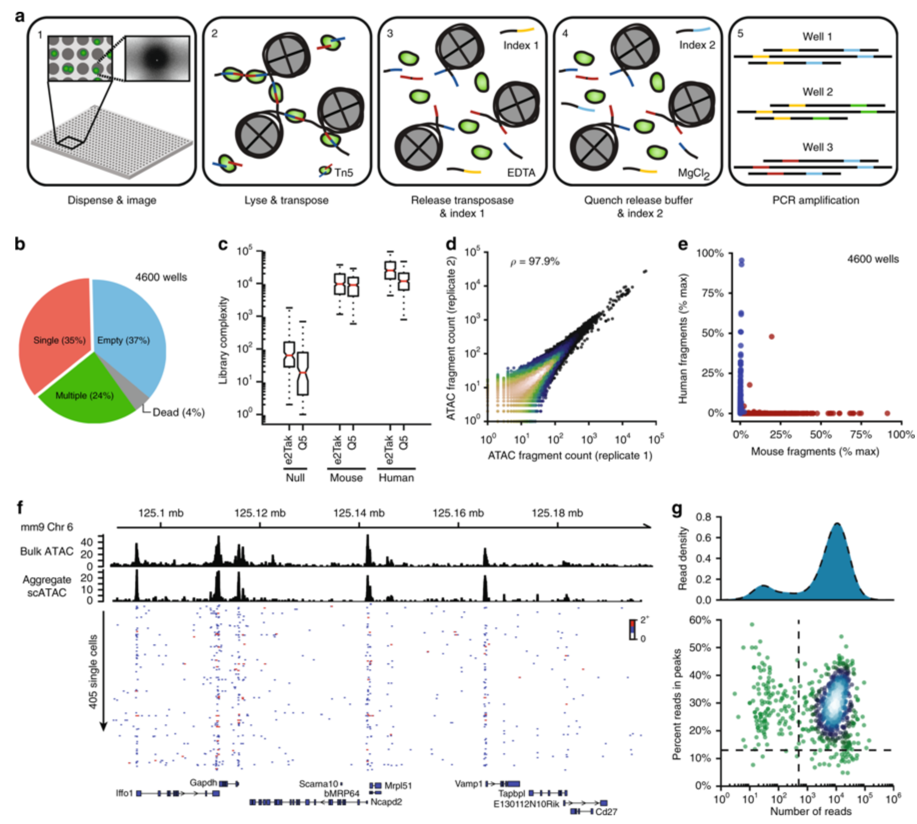
Anja Mezger, Sandy Klemm, Ishminder Mann, Kara Brower, Alain Mir, Magnolia Bostick, Andrew Farmer, Polly Fordyce, Sten Linnarsson & William Greenleaf (2018) “ High-throughput chromatin accessibility profiling at single-cell resolution. ” Nature Communications
High-throughput chromatin accessibility profiling at single-cell resolution
Here we develop a high-throughput single-cell ATAC-seq (assay for transposition of accessible chromatin) method to measure physical access to DNA in whole cells. Our approach integrates fluorescence imaging and addressable reagent deposition across a massively parallel (5184) nano-well array, yielding a nearly 20-fold improvement in throughput (up to ~1800 cells/chip, 4–5 h on-chip processing time) and library preparation cost (~81¢ per cell) compared to prior microfluidic implementations. We apply this method to measure regulatory variation in peripheral blood mononuclear cells (PBMCs) and show robust, de novo clustering of single cells by hematopoietic cell type.

Sarah Knight Denny, William James Greenleaf (2018) “ Linking RNA Sequence, Structure, and Function on Massively Parallel High-Throughput Sequencers .” Cold Spring Harbor Perspectives in Biology
Linking RNA Sequence, Structure, and Function on Massively Parallel High-Throughput Sequencers
High-throughput sequencing methods have revolutionized our ability to catalog the diversity of RNAs and RNA–protein interactions that can exist in our cells. However, the relationship between RNA sequence, structure, and function is enormously complex, demonstrating the need for methods that can provide quantitative thermodynamic and kinetic measurements of macromolecular interaction with RNA, at a scale commensurate with the sequence diversity of RNA. Here, we discuss a class of methods that extend the core functionality of DNA sequencers to enable high-throughput measurements of RNA folding and RNA–protein interactions. Topics discussed include a description of the method and multiple applications to RNA-binding proteins, riboswitch design and engineering, and RNA tertiary structure energetics.
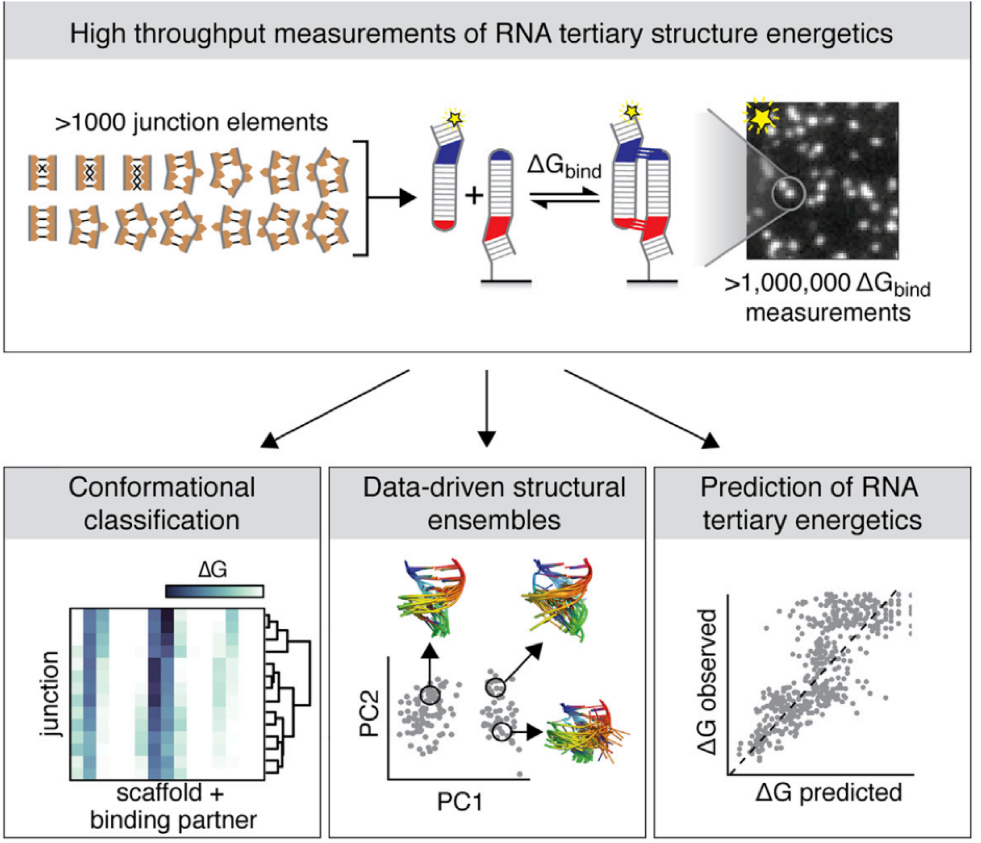
Sarah Knight Denny, Namita Bisaria, Joseph David Yesselman, Rhiju Das, Daniel Herschlag, William James Greenleaf (2018) “ High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding .” Cell
High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding
RNAs fold into defined tertiary structures to function in critical biological processes. While quantitative models can predict RNA secondary structure stability, we are still unable to predict the thermodynamic stability of RNA tertiary structure. Here, we probe conformational preferences of diverse RNA two-way junctions to develop a predictive model for the formation of RNA tertiary structure. We quantitatively measured tertiary assembly energetics of >1,000 of RNA junctions inserted in multiple structural scaffolds to generate a “thermodynamic fingerprint” for each junction. Thermodynamic fingerprints enabled comparison of junction conformational preferences, revealing principles for how sequence influences 3-dimensional conformations. Utilizing fingerprints of junctions with known crystal structures, we generated ensembles for related junctions that predicted their thermodynamic effects on assembly formation. This work reveals sequence-structure-energetic relationships in RNA, demonstrates the capacity for diverse compensation strategies within tertiary structures, and provides a path to quantitative modeling of RNA folding energetics based on “ensemble modularity.”
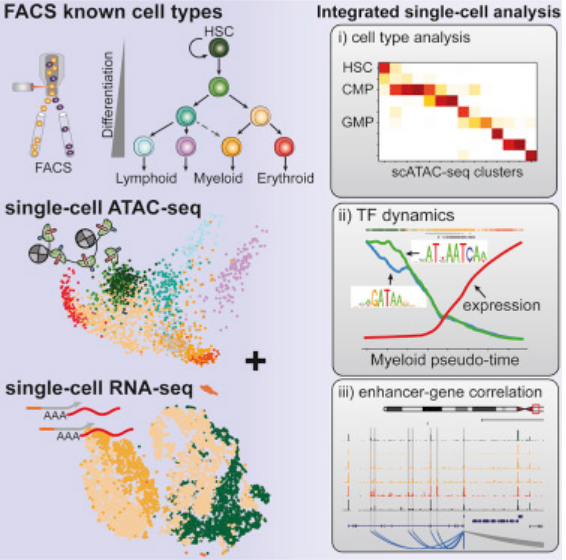
Jason D.Buenrostro, Ryan Corces, Caleb A.Lareau, Beijing Wu, Alicia N.Schep, Martin J.Aryee, Ravindra Majeti, Howard Y.Chang, William J.Greenleaf (2018) “Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation.” Cell
Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation
Human hematopoiesis involves cellular differentiation of multipotent cells into progressively more lineage-restricted states. While the chromatin accessibility landscape of this process has been explored in defined populations, single-cell regulatory variation has been hidden by ensemble averaging. We collected single-cell chromatin accessibility profiles across 10 populations of immunophenotypically defined human hematopoietic cell types and constructed a chromatin accessibility landscape of human hematopoiesis to characterize differentiation trajectories. We find variation consistent with lineage bias toward different developmental branches in multipotent cell types. We observe heterogeneity within common myeloid progenitors (CMPs) and granulocyte-macrophage progenitors (GMPs) and develop a strategy to partition GMPs along their differentiation trajectory. Furthermore, we integrated single-cell RNA sequencing (scRNA-seq) data to associate transcription factors to chromatin accessibility changes and regulatory elements to target genes through correlations of expression and regulatory element accessibility. Overall, this work provides a framework for integrative exploration of complex regulatory dynamics in a primary human tissue at single-cell resolution.
Dimitra Aggeli, Vlad O Karas, Nicholas A Sinnott-Armstrong, Vici Varghese, Robert W Shafer, William J Greenleaf, Gavin Sherlock (2018) “Diff-seq: A high throughput sequencing-based mismatch detection assay for DNA variant enrichment and discovery.” Nucleic Acids Research
Diff-seq: A high throughput sequencing-based mismatch detection assay for DNA variant enrichment and discovery
Much of the within species genetic variation is in the form of single nucleotide polymorphisms (SNPs), typically detected by whole genome sequencing (WGS) or microarray-based technologies. However, WGS produces mostly uninformative reads that perfectly match the reference, while microarrays require genome-specific reagents. We have developed Diff-seq, a sequencing-based mismatch detection assay for SNP discovery without the requirement for specialized nucleic-acid reagents. Diff-seq leverages the Surveyor endonuclease to cleave mismatched DNA molecules that are generated after cross-annealing of a complex pool of DNA fragments. Sequencing libraries enriched for Surveyor-cleaved molecules result in increased coverage at the variant sites. Diff-seq detected all mismatches present in an initial test substrate, with specific enrichment dependent on the identity and context of the variation. Application to viral sequences resulted in increased observation of variant alleles in a biologically relevant context. Diff-Seq has the potential to increase the sensitivity and efficiency of high-throughput sequencing in the detection of variation.
Graeme J. Gowans, Alicia N. Schep, Ka Man Wong, Devin A. King, William J. Greenleaf, Ashby J. Morrison (2018) “INO80 Chromatin Remodeling Coordinates Metabolic Homeostasis with Cell Division.” Cell Reports
INO80 Chromatin Remodeling Coordinates Metabolic Homeostasis with Cell Division
Adaptive survival requires the coordination of nutrient availability with expenditure of cellular resources. For example, in nutrient-limited environments, 50% of all S. cerevisiae genes synchronize and exhibit periodic bursts of expression in coordination with respiration and cell division in the yeast metabolic cycle (YMC). Despite the importance of metabolic and proliferative synchrony, the majority of YMC regulators are currently unknown. Here, we demonstrate that the INO80 chromatin-remodeling complex is required to coordinate respiration and cell division with periodic gene expression. Specifically, INO80 mutants have severe defects in oxygen consumption and promiscuous cell division that is no longer coupled with metabolic status. In mutant cells, chromatin accessibility of periodic genes, including TORC1-responsive genes, is relatively static, concomitant with severely attenuated gene expression. Collectively, these results reveal that the INO80 complex mediates metabolic signaling to chromatin to restrict proliferation to metabolically optimal states.
Maxwell R Mumbach, Ansuman T Satpathy, Evan A Boyle, Chao Dai, Benjamin G Gowen, Seung Woo Cho, Michelle L Nguyen, Adam J Rubin, Jeffrey M Granja, Katelynn R Kazane, Yuning Wei, Trieu Nguyen, Peyton G Greenside, M Ryan Corces, Josh Tycko, Dimitre R Simeonov, Nabeela Suliman, Rui Li, Jin Xu, Ryan A Flynn, Anshul Kundaje, Paul A Khavari, Alexander Marson, Jacob E Corn, Thomas Quertermous, William J Greenleaf & Howard Y Chang (2017) “Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements.” Nature Genetics
Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements
The challenge of linking intergenic mutations to target genes has limited molecular understanding of human diseases. Here we show that H3K27ac HiChIP generates high-resolution contact maps of active enhancers and target genes in rare primary human T cell subtypes and coronary artery smooth muscle cells. Differentiation of naive T cells into T helper 17 cells or regulatory T cells creates subtype-specific enhancer–promoter interactions, specifically at regions of shared DNA accessibility. These data provide a principled means of assigning molecular functions to autoimmune and cardiovascular disease risk variants, linking hundreds of noncoding variants to putative gene targets. Target genes identified with HiChIP are further supported by CRISPR interference and activation at linked enhancers, by the presence of expression quantitative trait loci, and by allele-specific enhancer loops in patient-derived primary cells. The majority of disease-associated enhancers contact genes beyond the nearest gene in the linear genome, leading to a fourfold increase in the number of potential target genes for autoimmune and cardiovascular diseases.
Dimitre R. Simeonov, Benjamin G. Gowen, Mandy Boontanrart, Theodore L. Roth, John D. Gagnon, Maxwell R. Mumbach, Ansuman T. Satpathy, Youjin Lee, Nicolas L. Bray, Alice Y. Chan, Dmytro S. Lituiev, Michelle L. Nguyen, Rachel E. Gate, Meena Subramaniam, Zhongmei Li, Jonathan M. Woo, Therese Mitros, Graham J. Ray, Gemma L. Curie, Nicki Naddaf, Julia S. Chu, Hong Ma, Eric Boyer, Frederic Van Gool, Hailiang Huang, Ruize Liu, Victoria R. Tobin, Kathrin Schumann, Mark J. Daly, Kyle K. Farh, K. Mark Ansel, Chun J. Ye, William J. Greenleaf, Mark S. Anderson, Jeffrey A. Bluestone, Howard Y. Chang, Jacob E. Corn & Alexander Marson (2017) “Discovery of stimulation-responsive immune enhancers with CRISPR activation.” Nature
Discovery of stimulation-responsive immune enhancers with CRISPR activation
The majority of genetic variants associated with common human diseases map to enhancers, non-coding elements that shape cell-type-specific transcriptional programs and responses to extracellular cues. Systematic mapping of functional enhancers and their biological contexts is required to understand the mechanisms by which variation in non-coding genetic sequences contributes to disease. Functional enhancers can be mapped by genomic sequence disruption, but this approach is limited to the subset of enhancers that are necessary in the particular cellular context being studied. We hypothesized that recruitment of a strong transcriptional activator to an enhancer would be sufficient to drive target gene expression, even if that enhancer was not currently active in the assayed cells. Here we describe a discovery platform that can identify stimulus-responsive enhancers for a target gene independent of stimulus exposure. We used tiled CRISPR activation (CRISPRa) to synthetically recruit a transcriptional activator to sites across large genomic regions (more than 100 kilobases) surrounding two key autoimmunity risk loci, CD69 and IL2RA. We identified several CRISPRa-responsive elements with chromatin features of stimulus-responsive enhancers, including an IL2RA enhancer that harbours an autoimmunity risk variant. Using engineered mouse models, we found that sequence perturbation of the disease-associated Il2ra enhancer did not entirely block Il2ra expression, but rather delayed the timing of gene activation in response to specific extracellular signals. Enhancer deletion skewed polarization of naive T cells towards a pro-inflammatory T helper (TH17) cell state and away from a regulatory T cell state. This integrated approach identifies functional enhancers and reveals how non-coding variation associated with human immune dysfunction alters context-specific gene programs.
M Ryan Corces, Alexandro E Trevino, Emily G Hamilton, Peyton G Greenside, Nicholas A Sinnott-Armstrong, Sam Vesuna, Ansuman T Satpathy, Adam J Rubin, Kathleen S Montine, Beijing Wu, Arwa Kathiria, Seung Woo Cho, Maxwell R Mumbach, Ava C Carter, Maya Kasowski, Lisa A Orloff, Viviana I Risca, Anshul Kundaje, Paul A Khavari, Thomas J Montine, William J Greenleaf & Howard Y Chang (2017) “An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues.” Nature Methods
An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues
We present Omni-ATAC, an improved ATAC-seq protocol for chromatin accessibility profiling that works across multiple applications with substantial improvement of signal-to-background ratio and information content. The Omni-ATAC protocol generates chromatin accessibility profiles from archival frozen tissue samples and 50-μm sections, revealing the activities of disease-associated DNA elements in distinct human brain structures. The Omni-ATAC protocol enables the interrogation of personal regulomes in tissue context and translational studies.
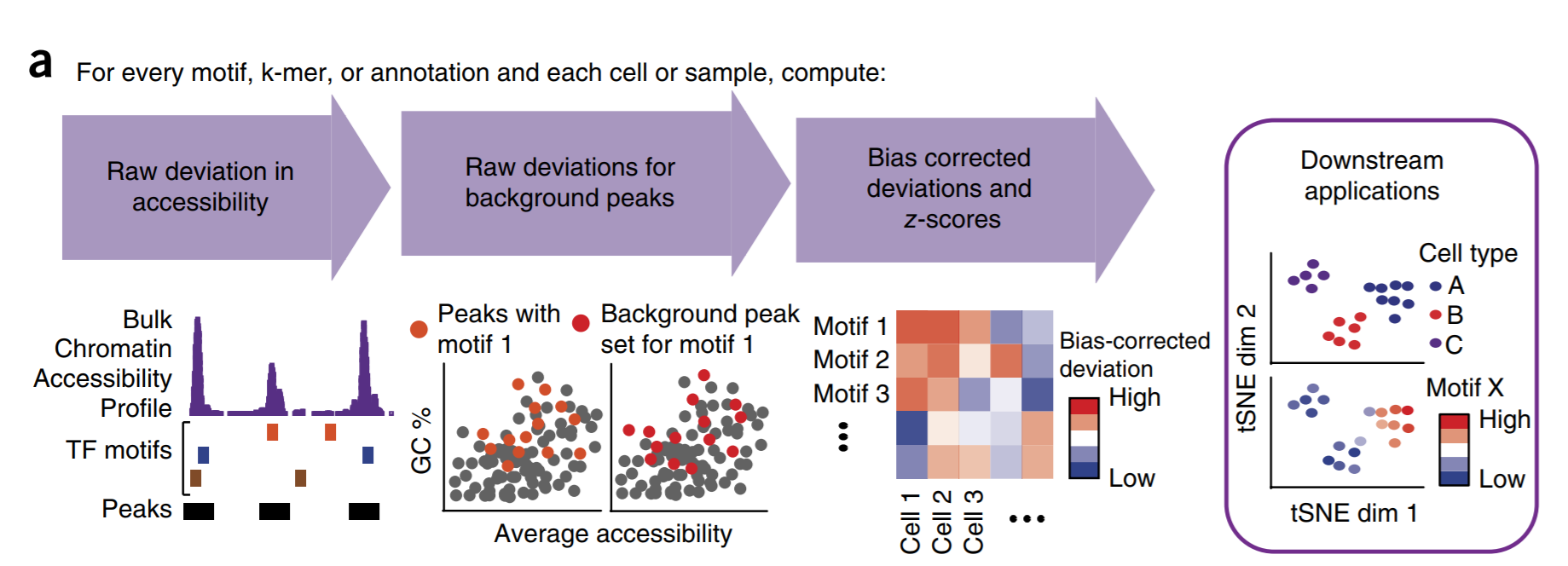
Alicia N Schep, Beijing Wu, Jason D Buenrostro & William J Greenleaf (2017) “chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data.” Nature Methods
chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data
Single-cell ATAC-seq (scATAC) yields sparse data that make conventional analysis challenging. We developed chromVAR (http://www.github.com/GreenleafLab/chromVAR), an R package for analyzing sparse chromatin-accessibility data by estimating gain or loss of accessibility within peaks sharing the same motif or annotation while controlling for technical biases. chromVAR enables accurate clustering of scATAC-seq profiles and characterization of known and de novo sequence motifs associated with variation in chromatin accessibility.
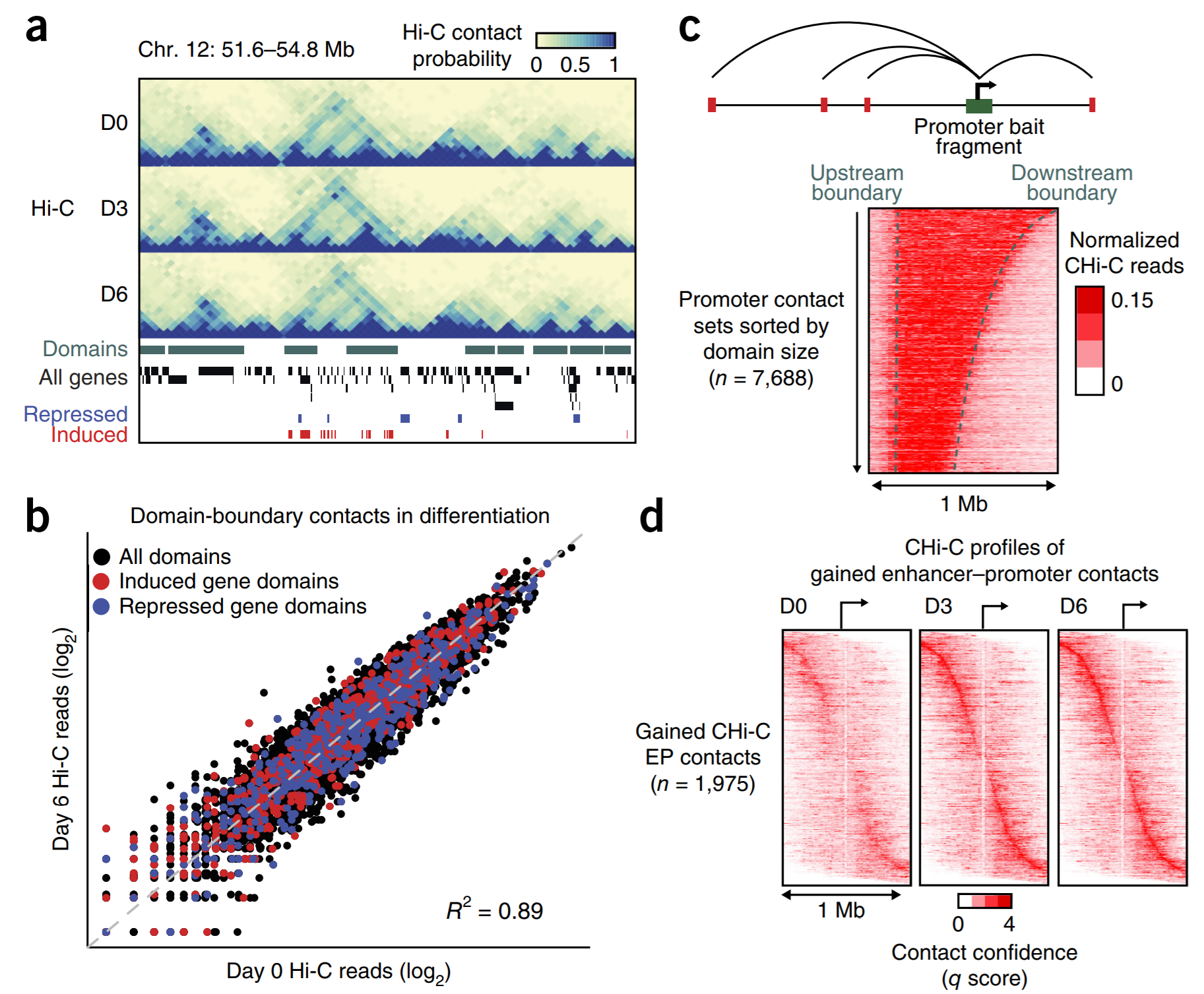
Adam J Rubin, Brook C Barajas, Mayra Furlan-Magaril, Vanessa Lopez-Pajares, Maxwell R Mumbach, Imani Howard, Daniel S Kim, Lisa D Boxer, Jonathan Cairns, Mikhail Spivakov, Steven W Wingett, Minyi Shi, Zhixin Zhao, William J Greenleaf, Anshul Kundaje, Michael Snyder, Howard Y Chang, Peter Fraser & Paul A Khavari. (2017) “Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation.” Nature Genetics
Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation
Chromosome conformation is an important feature of metazoan gene regulation1, 2; however, enhancer–promoter contact remodeling during cellular differentiation remains poorly understood3. To address this, genome-wide promoter capture Hi-C (CHi-C)1, 4 was performed during epidermal differentiation5. Two classes of enhancer–promoter contacts associated with differentiation-induced genes were identified. The first class ('gained') increased in contact strength during differentiation in concert with enhancer acquisition of the H3K27ac activation mark. The second class ('stable') were pre-established in undifferentiated cells, with enhancers constitutively marked by H3K27ac. The stable class was associated with the canonical conformation regulator cohesin, whereas the gained class was not, implying distinct mechanisms of contact formation and regulation. Analysis of stable enhancers identified a new, essential role for a constitutively expressed, lineage-restricted ETS-family transcription factor, EHF, in epidermal differentiation. Furthermore, neither class of contacts was observed in pluripotent cells, suggesting that lineage-specific chromatin structure is established in tissue progenitor cells and is further remodeled in terminal differentiation.

Marc P. Forrest, Hanwen Zhang, Winton Moy, Heather McGowan, Catherine Leites, Leonardo E. Dionisio, Zihui Xu, Jianxin Shi, Alan R. Sanders, William J. Greenleaf, Chad A. Cowan, Zhiping P. Pang, Pablo V. Gejman, Peter Penzes, Jubao Duan. (2017) “Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci..” Cell Stem Cell
Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci
Most disease variants lie within noncoding genomic regions, making their functional interpretation challenging. Because chromatin openness strongly influences transcriptional activity, we hypothesized that cell-type-specific open chromatin regions (OCRs) might highlight disease-relevant noncoding sequences. To investigate, we mapped global OCRs in neurons differentiating from hiPSCs, a cellular model for studying neurodevelopmental disorders such as schizophrenia (SZ). We found that the OCRs are highly dynamic and can stratify GWAS-implicated SZ risk variants. Of the more than 3,500 SZ-associated variants analyzed, we prioritized ∼100 putatively functional ones located in neuronal OCRs, including rs1198588, at a leading risk locus flanking MIR137. Excitatory neurons derived from hiPSCs with CRISPR/Cas9-edited rs1198588 or a rare proximally located SZ risk variant showed altered MIR137 expression, dendrite arborization, and synapse maturation. Our study shows that noncoding disease variants in OCRs can affect neurodevelopment, and that analysis of open chromatin regions can help prioritize functionally relevant noncoding variants identified by GWAS.

Kun Qu, Lisa C. Zaba, Ansuman T. Satpathy, Paul G. Giresi, Rui Li, Yonghao Jin, Randall Armstrong, Chen Jin, Nathalie Schmitt, Ziba Rahbar, Hideki Ueno, William J. Greenleaf, Youn H. Kim, Howard Y. Chang. (2017) “Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors.” Cancer Cell
Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors
Here, we define the landscape and dynamics of active regulatory DNA in cutaneous T cell lymphoma (CTCL) by ATAC-seq. Analysis of 111 human CTCL and control samples revealed extensive chromatin signatures that distinguished leukemic, host, and normal CD4+ T cells. We identify three dominant patterns of transcription factor (TF) activation that drive leukemia regulomes, as well as TF deactivations that alter host T cells in CTCL patients. Clinical response to histone deacetylase inhibitors (HDACi) is strongly associated with a concurrent gain in chromatin accessibility. HDACi causes distinct chromatin responses in leukemic and host CD4+ T cells, reprogramming host T cells toward normalcy. These results provide a foundational framework to study personal regulomes in human cancer and epigenetic therapy.
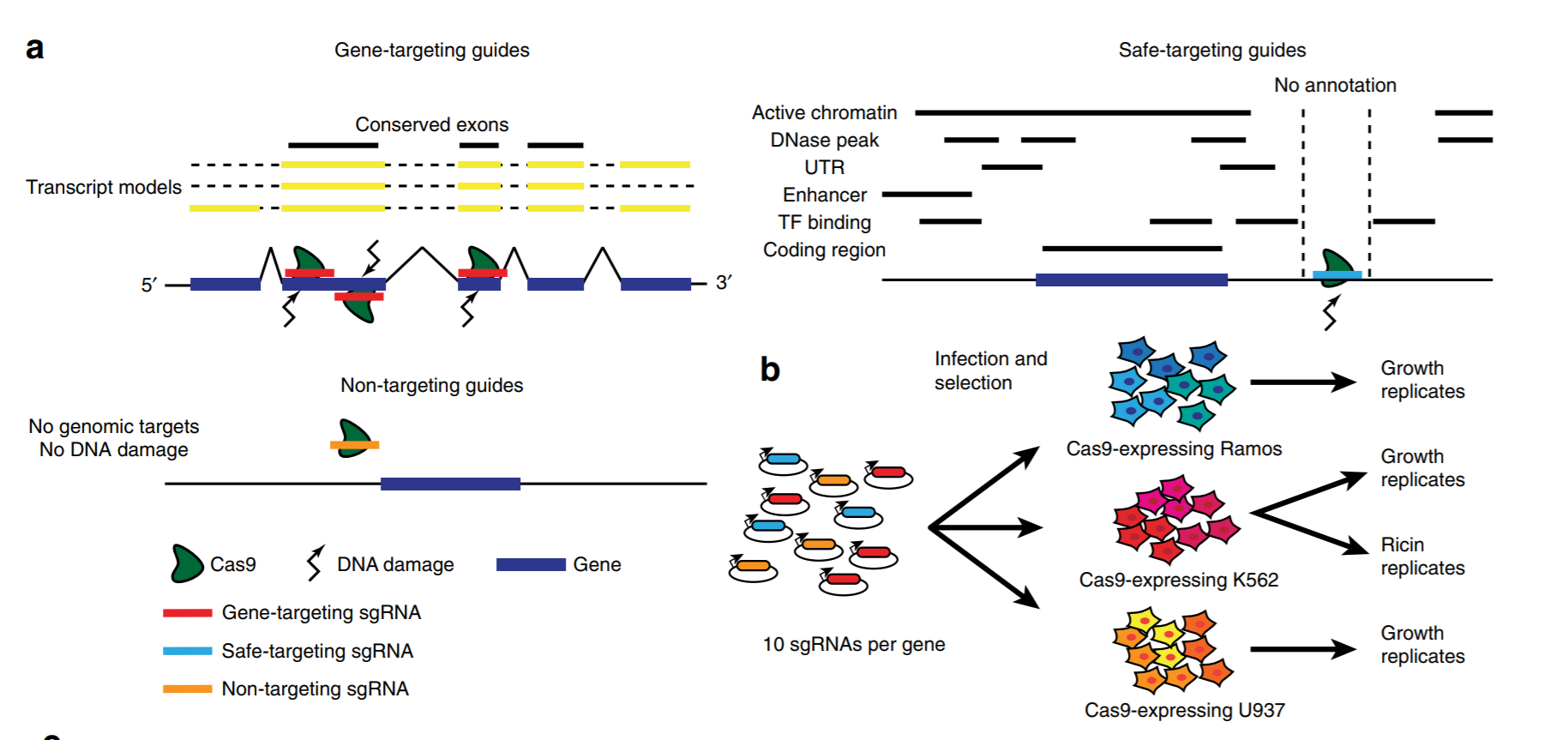
David W. Morgens,*, Michael Wainberg,*, Evan A. Boyle, Oana Ursu, Carlos L. Araya, C. Kimberly Tsui, Michael S. Haney, Gaelen T. Hess, Kyuho Han, Edwin E. Jeng, Amy Li, Michael P. Snyder, William J. Greenleaf, Anshul Kundaje & Michael C. Bassik.(2017) “Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screen” Nature Communications
Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens
CRISPR-Cas9 screens are powerful tools for high-throughput interrogation of genome function, but can be confounded by nuclease-induced toxicity at both on- and off-target sites, likely due to DNA damage. Here, to test potential solutions to this issue, we design and analyse a CRISPR-Cas9 library with 10 variable-length guides per gene and thousands of negative controls targeting non-functional, non-genic regions (termed safe-targeting guides), in addition to non-targeting controls. We find this library has excellent performance in identifying genes affecting growth and sensitivity to the ricin toxin. The safe-targeting guides allow for proper control of toxicity from on-target DNA damage. Using this toxicity as a proxy to measure off-target cutting, we demonstrate with tens of thousands of guides both the nucleotide position-dependent sensitivity to single mismatches and the reduction of off-target cutting using truncated guides. Our results demonstrate a simple strategy for high-throughput evaluation of target specificity and nuclease toxicity in Cas9 screens.
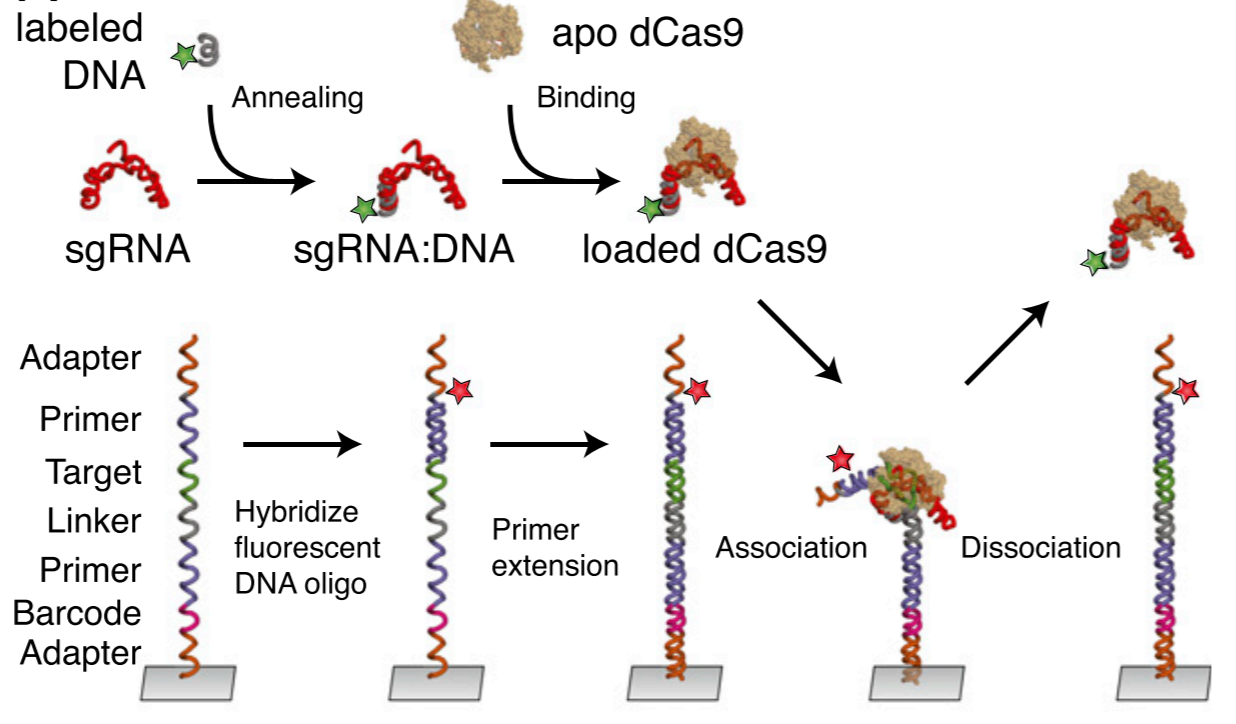
Evan A. Boyle, Johan O. L. Andreasson, Lauren M. Chircus, Samuel H. Sternberg, Michelle J. Wue, Chantal K. Guegler, Jennifer A. Doudna, and William J. Greenleaf.(2017) “High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding.” PNAS
High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding
The bacterial adaptive immune system CRISPR–Cas9 has been appropriated as a versatile tool for editing genomes, controlling gene expression, and visualizing genetic loci. To analyze Cas9’s ability to bind DNA rapidly and specifically, we generated multiple libraries of potential binding partners for measuring the kinetics of nuclease-dead Cas9 (dCas9) interactions. Using a massively parallel method to quantify protein–DNA interactions on a high-throughput sequencing flow cell, we comprehensively assess the effects of combinatorial mismatches between guide RNA (gRNA) and target nucleotides, both in the seed and in more distal nucleotides, plus disruption of the protospacer adjacent motif (PAM). We report two consequences of PAM-distal mismatches: reversal of dCas9 binding at long time scales, and synergistic changes in association kinetics when other gRNA–target mismatches are present. Together, these observations support a model for Cas9 specificity wherein gRNA–DNA mismatches at PAM-distal bases modulate different biophysical parameters that determine association and dissociation rates. The methods we present decouple aspects of kinetic and thermodynamic properties of the Cas9–DNA interaction and broaden the toolkit for investigating off-target binding behavior.
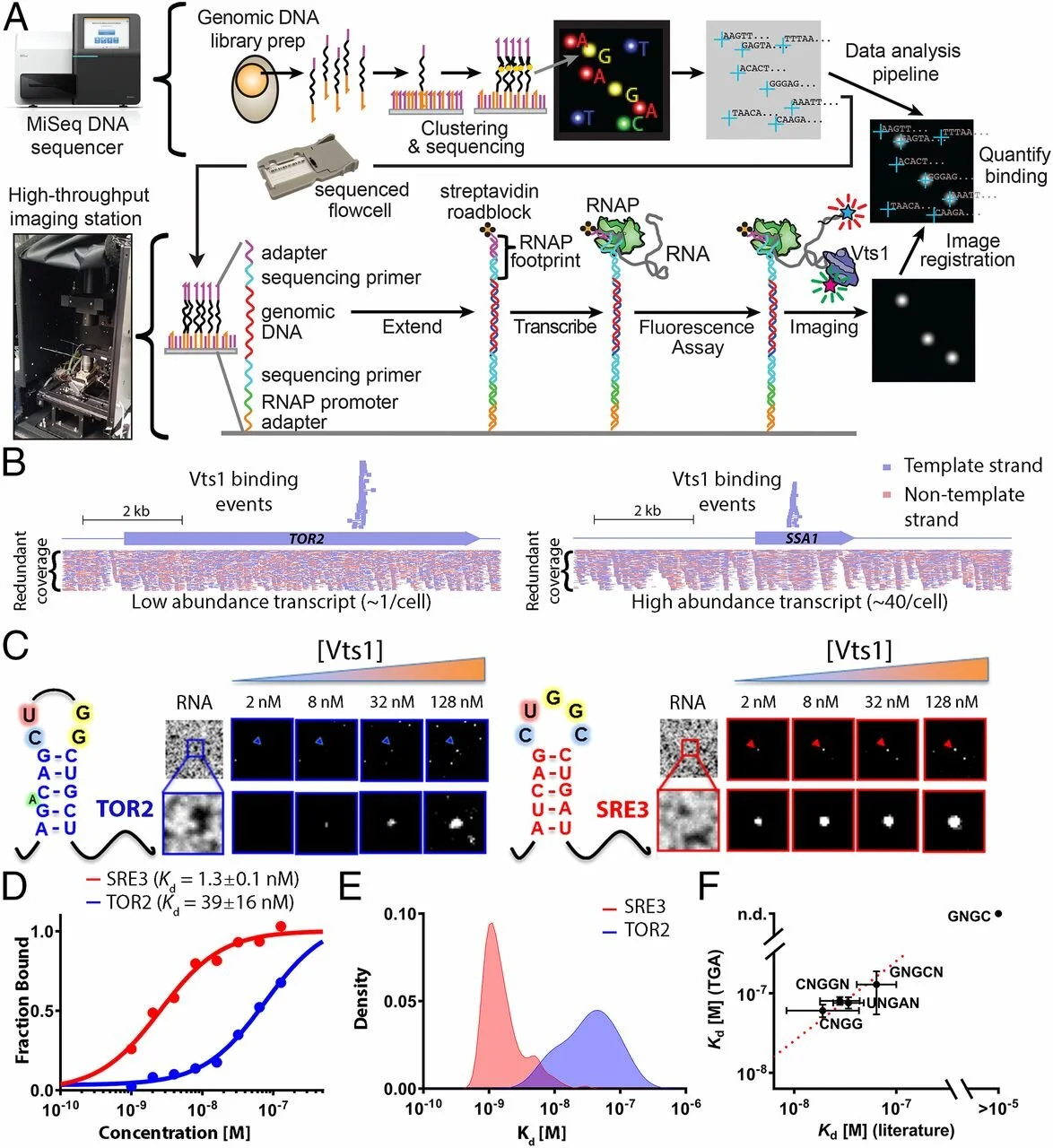
She R*, Chakravarty AK*, Layton CJ*, Chircus LM, Andreasson JOL, Damaraju N, McMahon PL, Buenrostro JD, Jarosz DF§, and Greenleaf WJ§. (2017) “Comprehensive and quantitative mapping of RNA–protein interactions across a transcribed eukaryotic genom” PNAS
Comprehensive and quantitative mapping of RNA-protein interactions across a transcribed eukaryotic genome
RNA-binding proteins (RBPs) control the fate of nearly every transcript in a cell. However, no existing approach for studying these posttranscriptional gene regulators combines transcriptome-wide throughput and biophysical precision. Here, we describe an assay that accomplishes this. Using commonly available hardware, we built a customizable, open-source platform that leverages the inherent throughput of Illumina technology for direct biophysical measurements. We used the platform to quantitatively measure the binding affinity of the prototypical RBP Vts1 for every transcript in the Saccharomyces cerevisiae genome. The scale and precision of these measurements revealed many previously unknown features of this well-studied RBP. Our transcribed genome array (TGA) assayed both rare and abundant transcripts with equivalent proficiency, revealing hundreds of low-abundance targets missed by previous approaches. These targets regulated diverse biological processes including nutrient sensing and the DNA damage response, and implicated Vts1 in de novo gene "birth." TGA provided single-nucleotide resolution for each binding site and delineated a highly specific sequence and structure motif for Vts1 binding. Changes in transcript levels in vts1Δ cells established the regulatory function of these binding sites. The impact of Vts1 on transcript abundance was largely independent of where it bound within an mRNA, challenging prevailing assumptions about how this RBP drives RNA degradation. TGA thus enables a quantitative description of the relationship between variant RNA structures, affinity, and in vivo phenotype on a transcriptome-wide scale. We anticipate that TGA will provide similarly comprehensive and quantitative insights into the function of virtually any RBP.
Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, Buenrostro JD, Weyand CM, Greenleaf WJ§, and Goronzy JJ§ (2017) “Epigenomics of human CD8 T cell differentiation and aging” Science Immunology
Epigenomics of human CD8 T cell differentiation and aging
The efficacy of the adaptive immune response declines markedly with age, but the cell-intrinsic mechanisms driving immune aging in humans remain poorly understood. Immune aging is characterized by a loss of self-renewing naïve cells and the accumulation of differentiated but dysfunctional cells within the CD8 T cell compartment. Using the assay for transposase-accessible chromatin using sequencing, we inferred that the transcription factor binding activities correlated with naïve and central and effector memory CD8 T cell states in young adults. Integrating our results with RNA sequencing, we identified transcription networks associated with CD8 T cell differentiation, with prominent roles implicated for BATF, ETS1, Eomes, and Sp1. Extending our analysis to aged humans, we found that the differences between the memory and naïve CD8 T cell subsets were largely preserved across age but that naïve and central memory cells from older individuals exhibited a shift toward more differentiated patterns of chromatin openness. In addition, aged naïve cells displayed a loss in chromatin accessibility at gene promoters, largely associated with a decrease in nuclear respiratory factor 1 (NRF1) binding. This shift was implicated in a marked drop-off in the ability of the aged naïve cells to transcribe respiratory chain genes, which may explain the reduced capacity of oxidative phosphorylation in older naïve cells. Our findings identify BATF- and NRF1-driven gene regulation as potential targets for delaying CD8 T cell aging and restoring function.
Litzenburger UM, Buenrostro JD, Wu B, Shen Y, Sheffield NC, Kathiria A, Greenleaf WJ, and Chang HY (2017) “Single-cell epigenomic variability reveals functional cancer heterogeneity” Genome Biology
Single-cell epigenomic variability reveals functional cancer heterogeneity
We develop a strategy to bridge the gap between measurement and function in single-cell epigenomics. Using single-cell chromatin accessibility and RNA-seq data in K562 leukemic cells, we identify the cell surface marker CD24 as co-varying with chromatin accessibility changes linked to GATA transcription factors in single cells. Fluorescence-activated cell sorting of CD24 high versus low cells prospectively isolated GATA1 and GATA2 high versus low cells. GATA high versus low cells express differential gene regulatory networks, differential sensitivity to the drug imatinib mesylate, and differential self-renewal capacity. Lineage tracing experiments show that GATA/CD24hi cells have the capability to rapidly reconstitute the heterogeneity within the entire starting population, suggesting that GATA expression levels drive a phenotypically relevant source of epigenomic plasticity.
Xu J, Carter AC, Gendrel A, Attia M, Loftus J, Greenleaf WJ, Tibshirani R, Heard E, and Chang HY (2017) “Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells” Nature Genetics
Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells
We developed an allele-specific assay for transposase-accessible chromatin with high-throughput sequencing (ATAC–seq) to genotype and profile active regulatory DNA across the genome. Using a mouse hybrid F1 system, we found that monoallelic DNA accessibility across autosomes was pervasive, developmentally programmed and composed of several patterns. Genetically determined accessibility was enriched at distal enhancers, but random monoallelically accessible (RAMA) elements were enriched at promoters and may act as gatekeepers of monoallelic mRNA expression. Allelic choice at RAMA elements was stable across cell generations and bookmarked through mitosis. RAMA elements in neural progenitor cells were biallelically accessible in embryonic stem cells but premarked with bivalent histone modifications; one allele was silenced during differentiation. Quantitative analysis indicated that allelic choice at the majority of RAMA elements is consistent with a stochastic process; however, up to 30% of RAMA elements may deviate from the expected pattern, suggesting a regulated or counting mechanism.
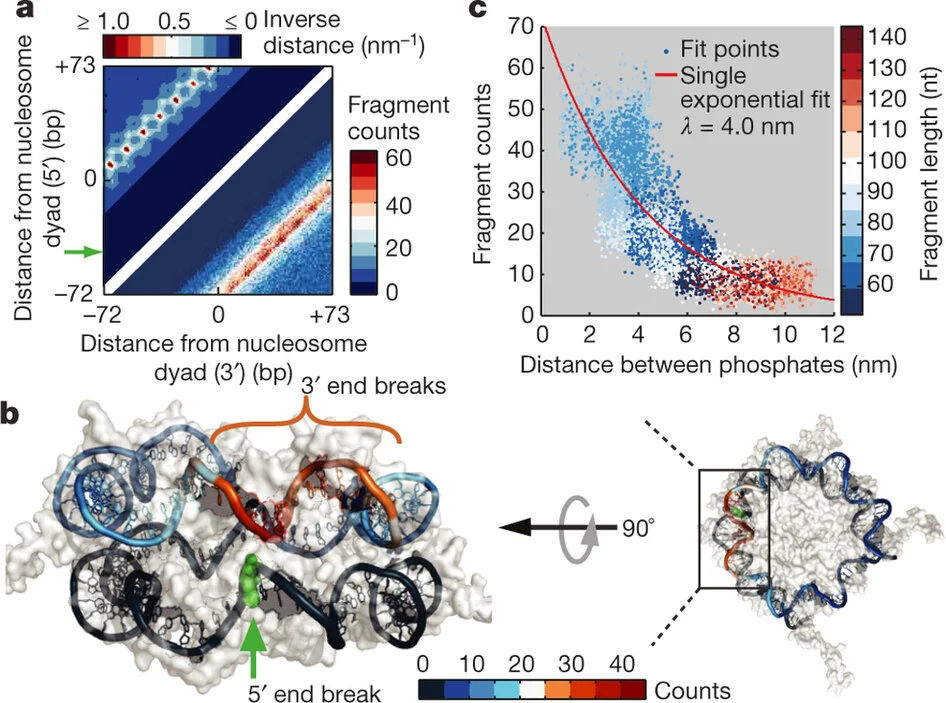
Risca VI, Denny SK, Straight AF, and Greenleaf WJ (2017) “Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping” Nature
Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping
Chromatin structure at the length scale encompassing local nucleosome–nucleosome interactions is thought to play a crucial role in regulating transcription and access to DNA1, 2, 3. However, this secondary structure of chromatin remains poorly understood compared with the primary structure of single nucleosomes or the tertiary structure of long-range looping interactions4. Here we report the first genome-wide map of chromatin conformation in human cells at the 1–3 nucleosome (50–500 bp) scale, obtained using ionizing radiation-induced spatially correlated cleavage of DNA with sequencing (RICC-seq) to identify DNA–DNA contacts that are spatially proximal. Unbiased analysis of RICC-seq signal reveals regional enrichment of DNA fragments characteristic of alternating rather than adjacent nucleosome interactions in tri-nucleosome units, particularly in H3K9me3-marked heterochromatin. We infer differences in the likelihood of nucleosome–nucleosome contacts among open chromatin, H3K27me3-marked, and H3K9me3-marked repressed chromatin regions. After calibrating RICC-seq signal to three-dimensional distances, we show that compact two-start helical fibre structures with stacked alternating nucleosomes are consistent with RICC-seq fragmentation patterns from H3K9me3-marked chromatin, while non-compact structures and solenoid structures are consistent with open chromatin. Our data support a model of chromatin architecture in intact interphase nuclei consistent with variable longitudinal compaction of two-start helical fibres.
Chen X, Shen Y, Draper W, Buenrostro JD, Litzenburger U, Cho SW, Satpathy AT, Carter AC, Ghosh RP, East-Seletsky A, Doudna JA, Greenleaf WJ, Liphardt JT, and Chang HY (2016) “ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing” Nature Methods
ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing
Spatial organization of the genome plays a central role in gene expression, DNA replication, and repair. But current epigenomic approaches largely map DNA regulatory elements outside of the native context of the nucleus. Here we report assay of transposase-accessible chromatin with visualization (ATAC-see), a transposase-mediated imaging technology that employs direct imaging of the accessible genome in situ, cell sorting, and deep sequencing to reveal the identity of the imaged elements. ATAC-see revealed the cell-type-specific spatial organization of the accessible genome and the coordinated process of neutrophil chromatin extrusion, termed NETosis. Integration of ATAC-see with flow cytometry enables automated quantitation and prospective cell isolation as a function of chromatin accessibility, and it reveals a cell-cycle dependence of chromatin accessibility that is especially dynamic in G1 phase. The integration of imaging and epigenomics provides a general and scalable approach for deciphering the spatiotemporal architecture of gene control.
Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016) “HiChIP: efficient and sensitive analysis of protein-directed genome architecture” Nature Methods
HiChIP: efficient and sensitive analysis of protein-directed genome architecture
Genome conformation is central to gene control but challenging to interrogate. Here we present HiChIP, a protein-centric chromatin conformation method. HiChIP improves the yield of conformation-informative reads by over 10-fold and lowers the input requirement over 100-fold relative to that of ChIA-PET. HiChIP of cohesin reveals multiscale genome architecture with greater signal-to-background ratios than those of in situ Hi-C.
Corces MR*, Buenrostro JD*, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R§, Chang HY§ (2016) “Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution” Nature Genetics
Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution
We define the chromatin accessibility and transcriptional landscapes in 13 human primary blood cell types that span the hematopoietic hierarchy. Exploiting the finding that the enhancer landscape better reflects cell identity than mRNA levels, we enable 'enhancer cytometry' for enumeration of pure cell types from complex populations. We identify regulators governing hematopoietic differentiation and further show the lineage ontogeny of genetic elements linked to diverse human diseases. In acute myeloid leukemia (AML), chromatin accessibility uncovers unique regulatory evolution in cancer cells with a progressively increasing mutation burden. Single AML cells exhibit distinctive mixed regulome profiles corresponding to disparate developmental stages. A method to account for this regulatory heterogeneity identified cancer-specific deviations and implicated HOX factors as key regulators of preleukemic hematopoietic stem cell characteristics. Thus, regulome dynamics can provide diverse insights into hematopoietic development and disease.
Denny SK*, Yang D*, Chuang C-H, Brady JJ, Lim JS, Grüner BM, Chiou S-H, Schep AN, Baral J, Hamard C, Antoine M., Wislez M, Kong CS, Connolly AJ, Park K-S, Sage J, Greenleaf WJ§, Winslow MM§ (2016) “Nfib promotes Metastasis through a Widespread Increase in Chromatin Accessibility” Cell
Nfib promotes Metastasis through a Widespread Increase in Chromatin Accessibility
Metastases are the main cause of cancer deaths, but the mechanisms underlying metastatic progression remain poorly understood. We have isolated pure populations of cancer cells from primary tumors and metastases from a genetically engineered mouse model of human small cell lung cancer (SCLC) to investigate the mechanisms that drive the metastatic spread of this lethal cancer. Genome-wide characterization of chromatin accessibility has revealed the opening of large numbers of distal regulatory elements across the genome during metastatic progression. These changes correlate with copy number amplification of the Nfib locus, and differentially accessible sites were highly enriched for Nfib transcription factor binding sites. Nfib is necessary and sufficient to increase chromatin accessibility at a large subset of the intergenic regions. Nfib promotes pro-metastatic neuronal gene expression programs and drives the metastatic ability of SCLC cells. The identification of widespread chromatin changes during SCLC progression reveals an unexpected global reprogramming during metastatic progression.
Araya CL*, Cenik C*, Reuter JA, Kiss G, Pande VS, Snyder MP, Greenleaf WJ. (2015) "Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations." Nature Genetics
Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations
Cancer sequencing studies have primarily identified cancer driver genes by the accumulation of protein-altering mutations. An improved method would be annotation independent, sensitive to unknown distributions of functions within proteins and inclusive of noncoding drivers. We employed density-based clustering methods in 21 tumor types to detect variably sized significantly mutated regions (SMRs). SMRs reveal recurrent alterations across a spectrum of coding and noncoding elements, including transcription factor binding sites and untranslated regions mutated in up to ~15% of specific tumor types. SMRs demonstrate spatial clustering of alterations in molecular domains and at interfaces, often with associated changes in signaling. Mutation frequencies in SMRs demonstrate that distinct protein regions are differentially mutated across tumor types, as exemplified by a linker region of PIK3CA in which biophysical simulations suggest that mutations affect regulatory interactions. The functional diversity of SMRs underscores both the varied mechanisms of oncogenic misregulation and the advantage of functionally agnostic driver identification.
Viviana I. Risca, William J. Greenleaf (2015) “Beyond the Linear Genome: Paired-End Sequencing as a Biophysical Tool” Trends in Cell Biology
[Review] Beyond the Linear Genome: Paired-End Sequencing as a Biophysical Tool
Paired-end sequencing has enabled a variety of new methods for high-throughput interrogation of both genome structure and chromatin architecture. Here, we discuss how the paired-end paradigm can be used to interpret sequencing data as biophysical measurements of in vivo chromatin structure that report on single molecules in single cells.
Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. (2015) "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions." Genome Research
Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions
Transcription factors canonically bind nucleosome-free DNA, making the positioning of nucleosomes within regulatory regions crucial to the regulation of gene expression. Using the assay of transposase accessible chromatin (ATAC-seq), we observe a highly structured pattern of DNA fragment lengths and positions around nucleosomes in Saccharomyces cerevisiae, and use this distinctive two-dimensional nucleosomal “fingerprint” as the basis for a new nucleosome-positioning algorithm called NucleoATAC. We show that NucleoATAC can identify the rotational and translational positions of nucleosomes with up to base-pair resolution and provide quantitative measures of nucleosome occupancy in S. cerevisiae, Schizosaccharomyces pombe, and human cells. We demonstrate the application of NucleoATAC to a number of outstanding problems in chromatin biology, including analysis of sequence features underlying nucleosome positioning, promoter chromatin architecture across species, identification of transient changes in nucleosome occupancy and positioning during a dynamic cellular response, and integrated analysis of nucleosome occupancy and transcription factor binding.
Kun Qu, Lisa C. Zaba, Paul G. Giresi, Rui Li, Michelle Longmire, Youn H. Kim, William J. Greenleaf, Howard Y. Chang (2015) “Individuality and Variation of Personal Regulomes in Primary Human T Cells“ Cell
Individuality and Variation of Personal Regulomes in Primary Human T Cells
Here, we survey variation and dynamics of active regulatory elements genome-wide using longitudinal samples from human individuals. We applied Assay of Transposase Accessible Chromatin with sequencing (ATAC-seq) to map chromatin accessibility in primary CD4+ T cells isolated from standard blood draws from 12 healthy volunteers over time, from cancer patients, and during T-cell activation. Over 4,000 predicted regulatory elements (7.2%) showed reproducible variation in accessibility between individuals. Gender was the most significant attributable source of variation. ATAC-seq revealed previously undescribed elements that escape X chromosome inactivation and predicted gender-specific gene regulatory networks across autosomes, which coordinately affect genes with immune function. Noisy regulatory elements with personal variation in accessibility are significantly enriched for autoimmune disease loci. Over one third of regulome variation lacked genetic variation in cis, suggesting contributions from environmental or epigenetic factors. These results refine concepts of human individuality and provide a foundational reference for comparing disease-associated regulomes.
Itay Maza, Inbal Caspi, Asaf Zviran, Elad Chomsky, Yoach Rais, Sergey Viukov, Shay Geula, Jason D Buenrostro, Leehee Weinberger, Vladislav Krupalnik, Suhair Hanna, Mirie Zerbib, James R Dutton, William J Greenleaf, Rada Massarwa, Noa Novershtern, Jacob H Hanna (2015) “Transient acquisition of pluripotency during somatic cell transdifferentiation with iPsC reprogramming factors” Nature Biotechnology
Transient acquisition of pluripotency during somatic cell transdifferentiation with iPSC reprogramming factors
Somatic cells can be transdifferentiated to other cell types without passing through a pluripotent state by ectopic expression of appropriate transcription factors. Recent reports have proposed an alternative transdifferentiation method in which fibroblasts are directly converted to various mature somatic cell types by brief expression of the induced pluripotent stem cell (iPSC) reprogramming factors Oct4, Sox2, Klf4 and c-Myc (OSKM) followed by cell expansion in media that promote lineage differentiation. Here we test this method using genetic lineage tracing for expression of endogenous Nanog and Oct4 and for X chromosome reactivation, as these events mark acquisition of pluripotency. We show that the vast majority of reprogrammed cardiomyocytes or neural stem cells obtained from mouse fibroblasts by OSKM-induced 'transdifferentiation' pass through a transient pluripotent state, and that their derivation is molecularly coupled to iPSC formation mechanisms. Our findings underscore the importance of defining trajectories during cell reprogramming by various methods.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. (2015) "Single-cell chromatin accessibility reveals principles of regulatory variation." Nature
Single-cell chromatin accessibility reveals principles of regulatory variation
Cell-to-cell variation is a universal feature of life that affects a wide range of biological phenomena, from developmental plasticity to tumour heterogeneity. Although recent advances have improved our ability to document cellular phenotypic variation, the fundamental mechanisms that generate variability from identical DNA sequences remain elusive. Here we reveal the landscape and principles of mammalian DNA regulatory variation by developing a robust method for mapping the accessible genome of individual cells by assay for transposase-accessible chromatin using sequencing (ATAC-seq) integrated into a programmable microfluidics platform. Single-cell ATAC-seq (scATAC-seq) maps from hundreds of single cells in aggregate closely resemble accessibility profiles from tens of millions of cells and provide insights into cell-to-cell variation. Accessibility variance is systematically associated with specific trans-factors and cis-elements, and we discover combinations of trans-factors associated with either induction or suppression of cell-to-cell variability. We further identify sets of trans-factors associated with cell-type-specific accessibility variance across eight cell types. Targeted perturbations of cell cycle or transcription factor signalling evoke stimulus-specific changes in this observed variability. The pattern of accessibility variation in cis across the genome recapitulates chromosome compartments de novo, linking single-cell accessibility variation to three-dimensional genome organization. Single-cell analysis of DNA accessibility provides new insight into cellular variation of the ‘regulome’.
Risca VI, Greenleaf WJ. (2015) "Unraveling the 3D genome: genomics tools for multiscale exploration." Trends in Genetics
[Review] Unraveling the 3D genome: genomics tools for multiscale exploration
A decade of rapid method development has begun to yield exciting insights into the 3D architecture of the metazoan genome and the roles it may play in regulating transcription. Here we review core methods and new tools in the modern genomicist's toolbox at three length scales, ranging from single base pairs to megabase-scale chromosomal domains, and discuss the emerging picture of the 3D genome that these tools have revealed. Blind spots remain, especially at intermediate length scales spanning a few nucleosomes, but thanks in part to new technologies that permit targeted alteration of chromatin states and time-resolved studies, the next decade holds great promise for hypothesis-driven research into the mechanisms that drive genome architecture and transcriptional regulation.

Jason D. Buenrostro, Beijing Wu, Howard Y. Chang, William J. Greenleaf (2015) “ATAC-seq: A Method for Assaying UNIT 21.29 Chromatin Accessibility Genome-Wide” Current Protocols in Molecular Biology
ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide
This unit describes Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq), a method for mapping chromatin accessibility genome-wide. This method probes DNA accessibility with hyperactive Tn5 transposase, which inserts sequencing adapters into accessible regions of chromatin. Sequencing reads can then be used to infer regions of increased accessibility, as well as to map regions of transcription-factor binding and nucleosome position. The method is a fast and sensitive alternative to DNase-seq for assaying chromatin accessibility genome-wide, or to MNase-seq for assaying nucleosome positions in accessible regions of the genome.
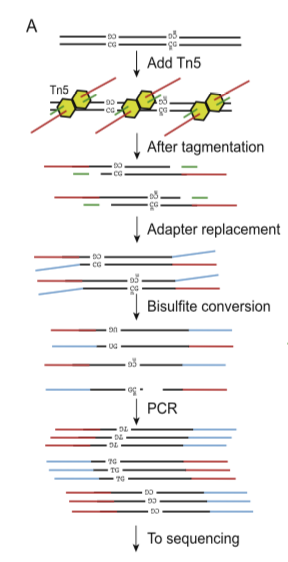
William J. Greenleaf (2015). “Assaying the epigenome in limited numbers of cells” Methods
Assaying the epigenome in limited numbers of cells
Spectacular advances in the throughput of DNA sequencing have allowed genome-wide analysis of epigenetic features such as methylation, nucleosome position and post-translational modification, chromatin accessibility and connectivity, and transcription factor binding. However, for rare or precious biological samples, input requirements of many of these methods limit their application. In this review we discuss recent advances for low-input genome-wide analysis of chromatin immunoprecipitation, methylation, DNA accessibility, and chromatin conformation.
Shin-Heng Chiou, Caroline Kim-Kiselak, Viviana I. Risca, Megan K. Heimann, Chen-Hua Chuang, Aurora A. Burds, William J. Greenleaf, Tyler E. Jacks, David M. Feldser, Monte M. Winslow (2014) “A Conditional System to Specifically Link Disruption of Protein-Coding Function with Reporter Expression in Mice” Cell Reports
A Conditional System to Specifically Link Disruption of Protein-Coding Function with Reporter Expression in Mice
Transcription by RNA polymerase (RNAP) is interrupted by pauses that play diverse regulatory roles. Although individual pauses have been studied in vitro, the determinants of pauses in vivo and their distribution throughout the bacterial genome remain unknown. Using nascent transcript sequencing we identify a 16-nt consensus pause sequence in E. coli that accounts for known regulatory pause sites as well as ~20,000 new in vivo pause sites. In vitro single-molecule and ensemble analyses demonstrate that these pauses result from RNAP/nucleic-acid interactions that inhibit next-nucleotide addition. The consensus sequence also leads to pausing by RNAPs from diverse lineages and is enriched at translation start sites in both E. coli and B. subtilis. Our results thus reveal a conserved mechanism unifying known and newly identified pause events.
Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ*, Landick R*, Weissman JS*. (2014) "A Pause Sequence Enriched at Translation Start Sites Drives Transcription Dynamics in Vivo." Science
A Pause Sequence Enriched at Translation Start Sites Drives Transcription Dynamics in Vivo
Transcription by RNA polymerase (RNAP) is interrupted by pauses that play diverse regulatory roles. Although individual pauses have been studied in vitro, the determinants of pauses in vivo and their distribution throughout the bacterial genome remain unknown. Using nascent transcript sequencing we identify a 16-nt consensus pause sequence in E. coli that accounts for known regulatory pause sites as well as ~20,000 new in vivo pause sites. In vitro single-molecule and ensemble analyses demonstrate that these pauses result from RNAP/nucleic-acid interactions that inhibit next-nucleotide addition. The consensus sequence also leads to pausing by RNAPs from diverse lineages and is enriched at translation start sites in both E. coli and B. subtilis. Our results thus reveal a conserved mechanism unifying known and newly identified pause events.
Buenrostro JD*, Araya CL*, Chircus LM, Layton CJ, Chang HY, Snyder MP, and Greenleaf WJ. (2014) "Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes." Nature Biotechnology
Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes
RNA-protein interactions drive fundamental biological processes and are targets for molecular engineering, yet quantitative and comprehensive understanding of the sequence determinants of affinity remains limited. Here we repurpose a high-throughput sequencing instrument to quantitatively measure binding and dissociation of a fluorescently labeled protein to >10^7 RNA targets generated on a flow-cell surface by in situ transcription and intermolecular tethering of RNA to DNA. Studying the MS2 coat protein, we decompose the binding energy contributions from primary and secondary RNA structure, finding that differences in affinity are often driven by sequence-specific changes in both association and dissociation rates. By analyzing the biophysical constraints and modeling mutational paths describing the molecular evolution of MS2 from low- to high-affinity hairpins, we quantify widespread molecular epistasis and a long-hypothesized, structure-dependent preference for G:U base pairs over C:A intermediates in evolutionary trajectories. Our results suggest that quantitative analysis of RNA on a massively parallel array (RNA-MaP) provides generalizable insight into the biophysical basis and evolutionary consequences of sequence-function relationships.
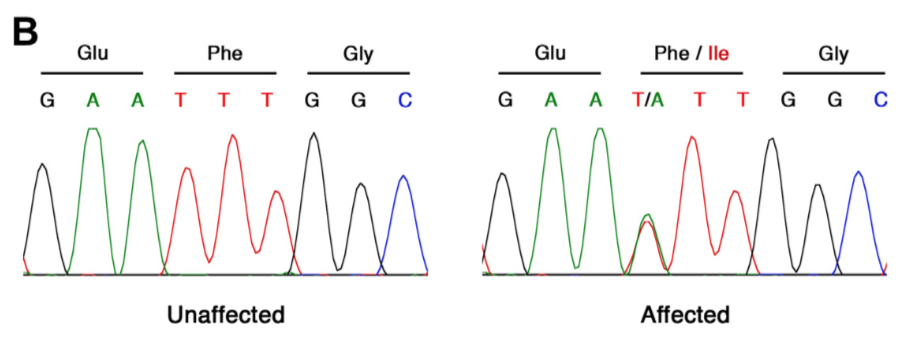
Julien Couthouis, Alya R. Raphael, Carly Siskind, Andrew R. Findlay, Jason D. Buenrostro, William J. Greenleaf, Hannes Vogel, John W. Day, Kevin M. Flanigan, Aaron D. Gitler. (2014) “Exome sequencing identifies a DNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophy” Neuromuscular Disorders
Exome sequencing identifies a DNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophy
Limb-girdle muscular dystrophy primarily affects the muscles of the hips and shoulders (the "limb-girdle" muscles), although it is a heterogeneous disorder that can present with varying symptoms. There is currently no cure. We sought to identify the genetic basis of limb-girdle muscular dystrophy type 1 in an American family of Northern European descent using exome sequencing. Exome sequencing was performed on DNA samples from two affected siblings and one unaffected sibling and resulted in the identification of eleven candidate mutations that co-segregated with the disease. Notably, this list included a previously reported mutation in DNAJB6, p.Phe89Ile, which was recently identified as a cause of limb-girdle muscular dystrophy type 1D. Additional family members were Sanger sequenced and the mutation in DNAJB6 was only found in affected individuals. Subsequent haplotype analysis indicated that this DNAJB6 p.Phe89Ile mutation likely arose independently of the previously reported mutation. Since other published mutations are located close by in the G/F domain of DNAJB6, this suggests that the area may represent a mutational hotspot. Exome sequencing provided an unbiased and effective method for identifying the genetic etiology of limb-girdle muscular dystrophy type 1 in a previously genetically uncharacterized family. This work further confirms the causative role of DNAJB6 mutations in limb-girdle muscular dystrophy type 1D.

William J Greenleaf, Arend Sidow (2014) “The future of sequencing: convergence of intelligent design and market Darwinism” Genome Biology
The future of sequencing: convergence of intelligent design and market Darwinism
A report on the Advances in Genome Biology and Technology meeting held in Marco Island, Florida, USA, on February 12–15, 2014.
Meredith L. Carpenter, Jason D. Buenrostro, Cristina Valdiosera, Hannes Schroeder, Morten E. Allentoft, Martin Sikora, Morten Rasmussen, Simon Gravel, Sonia Guillén, Georgi Nekhrizov, Krasimir Leshtakov, Diana Dimitrova, Nikola Theodossiev, Davide Pettener, Donata Luiselli, Karla Sandoval, Andre´s Moreno-Estrada, Yingrui Li, Jun Wang, M. Thomas P. Gilbert, Eske Willerslev, William J. Greenleaf, Carlos D. Bustamante (2013) “Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries” American Journal of Human Genetics
Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries
Most ancient specimens contain very low levels of endogenous DNA, precluding the shotgun sequencing of many interesting samples because of cost. Ancient DNA (aDNA) libraries often contain <1% endogenous DNA, with the majority of sequencing capacity taken up by environmental DNA. Here we present a capture-based method for enriching the endogenous component of aDNA sequencing libraries. By using biotinylated RNA baits transcribed from genomic DNA libraries, we are able to capture DNA fragments from across the human genome. We demonstrate this method on libraries created from four Iron Age and Bronze Age human teeth from Bulgaria, as well as bone samples from seven Peruvian mummies and a Bronze Age hair sample from Denmark. Prior to capture, shotgun sequencing of these libraries yielded an average of 1.2% of reads mapping to the human genome (including duplicates). After capture, this fraction increased substantially, with up to 59% of reads mapped to human and enrichment ranging from 6- to 159-fold. Furthermore, we maintained coverage of the majority of regions sequenced in the precapture library. Intersection with the 1000 Genomes Project reference panel yielded an average of 50,723 SNPs (range 3,062–147,243) for the postcapture libraries sequenced with 1 million reads, compared with 13,280 SNPs (range 217–73,266) for the precapture libraries, increasing resolution in population genetic analyses. Our whole-genome capture approach makes it less costly to sequence aDNA from specimens containing very low levels of endogenous DNA, enabling the analysis of larger numbers of samples.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ. (2013) "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position." Nature Methods
Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position
We describe an assay for transposase-accessible chromatin using sequencing (ATAC-seq), based on direct in vitro transposition of sequencing adaptors into native chromatin, as a rapid and sensitive method for integrative epigenomic analysis. ATAC-seq captures open chromatin sites using a simple two-step protocol with 500-50,000 cells and reveals the interplay between genomic locations of open chromatin, DNA-binding proteins, individual nucleosomes and chromatin compaction at nucleotide resolution. We discovered classes of DNA-binding factors that strictly avoided, could tolerate or tended to overlap with nucleosomes. Using ATAC-seq maps of human CD4(+) T cells from a proband obtained on consecutive days, we demonstrated the feasibility of analyzing an individual's epigenome on a timescale compatible with clinical decision-making.
Yongfan Men, Yusi Fu, Zitian Chen, Peter A. Sims, William J. Greenleaf, Yanyi Huang (2012) “Digital Polymerase Chain Reaction in an Array of Femtoliter Polydimethylsiloxane Microreactors” Analytical Chemistry
Digital polymerase chain reaction in an array of femtoliter polydimethylsiloxane microreactors
We developed a simple, compact microfluidic device to perform high dynamic-range digital polymerase chain reaction (dPCR) in an array of isolated 36-femtoliter microreactors. The density of the microreactors exceeded 20000/mm(2). This device, made from polydimethylsiloxane (PDMS), allows the samples to be loaded into all microreactors simultaneously. The microreactors are completely sealed through the deformation of a PDMS membrane. The small volume of the microreactors ensures a compact device with high reaction efficiency and low reagent and sample consumption. Future potential applications of this platform include multicolor dPCR and massively parallel dPCR for next generation sequencing library preparation.
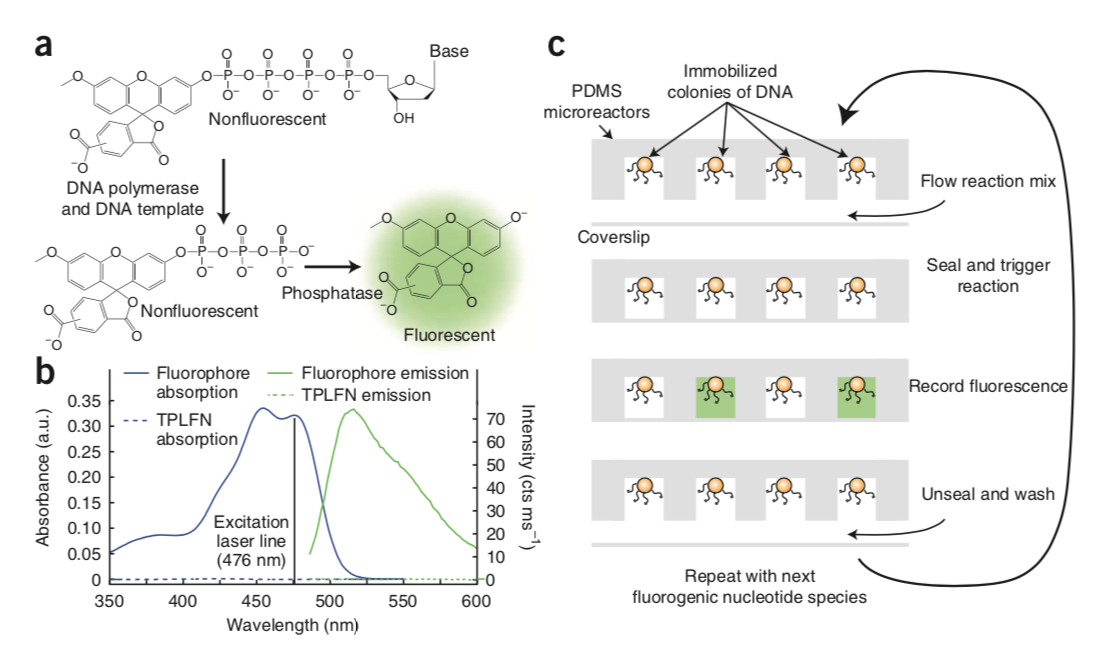
Peter A Sims, William J Greenleaf, Haifeng Duan, X Sunney Xie. (2011) “Fluorogenic DNA sequencing in PDMS microreactors” Nature Methods
Fluorogenic DNA sequencing in PDMS microreactors
We developed a multiplex sequencing-by-synthesis method combining terminal phosphate-labeled fluorogenic nucleotides (TPLFNs) and resealable polydimethylsiloxane (PDMS) microreactors. In the presence of phosphatase, primer extension by DNA polymerase using nonfluorescent TPLFNs generates fluorophores, which are confined in the microreactors and detected. We immobilized primed DNA templates in the microreactors, then sequentially introduced one of the four identically labeled TPLFNs, sealed the microreactors and recorded a fluorescence image after template-directed primer extension. With cycle times of <10 min, we demonstrate 30 base reads with ∼99% raw accuracy. Our 'fluorogenic pyrosequencing' offers benefits of pyrosequencing, such as rapid turnaround, one-color detection and generation of native DNA, along with high detection sensitivity and simplicity of parallelization because simultaneous real-time monitoring of all microreactors is not required.
Braulio Gutiérrez-Medina, Johan O.L. Andreasson, William J. Greenleaf, Arthur LaPorta, Steven M.Block (2010) “An Optical Apparatus for Rotation and Trapping” Methods in Enzymology
[Chapter] An optical apparatus for rotation and trapping
We present details of the design, construction, and testing of a single-beam optical tweezers apparatus capable of measuring and exerting torque, as well as force, on microfabricated, optically anisotropic particles (an "optical torque wrench"). The control of angular orientation is achieved by rotating the linear polarization of a trapping laser with an electro-optic modulator (EOM), which affords improved performance over previous designs. The torque imparted to the trapped particle is assessed by measuring the difference between left- and right-circular components of the transmitted light, and constant torque is maintained by feeding this difference signal back into a custom-designed electronic servo loop. The limited angular range of the EOM (+/-180 degrees ) is extended by rapidly reversing the polarization once a threshold angle is reached, enabling the torque clamp to function over unlimited, continuous rotations at high bandwidth. In addition, we developed particles suitable for rotation in this apparatus using microfabrication techniques. Altogether, the system allows for the simultaneous application of forces (approximately 0.1-100 pN) and torques (approximately 1-10,000 pN nm) in the study of biomolecules. As a proof of principle, we demonstrate how our instrument can be used to study the supercoiling of single DNA molecules.
Greenleaf WJ*, Frieda KL, Foster, DAN, Woodside MT*, and Block SM. (2008) “Direct observation of hierarchical folding in single riboswitch aptamers.” Science, 319:630-633.
Direct observation of hierarchical folding in single riboswitch aptamers
Riboswitches regulate genes through structural changes in ligand-binding RNA aptamers. With the use of an optical-trapping assay based on in situ transcription by a molecule of RNA polymerase, single nascent RNAs containing pbuE adenine riboswitch aptamers were unfolded and refolded. Multiple folding states were characterized by means of both force-extension curves and folding trajectories under constant force by measuring the molecular contour length, kinetics, and energetics with and without adenine. Distinct folding steps correlated with the formation of key secondary or tertiary structures and with ligand binding. Adenine-induced stabilization of the weakest helix in the aptamer, the mechanical switch underlying regulatory action, was observed directly. These results provide an integrated view of hierarchical folding in an aptamer, demonstrating how complex folding can be resolved into constituent parts, and supply further insights into tertiary structure formation.
Matthew H. Larson, William J. Greenleaf, Robert Landick, Steven M. Block (2008) “Applied Force Reveals Mechanistic and Energetic Details of Transcription Termination” Cell
Applied force reveals mechanistic and energetic details of transcription termination
Transcription termination by bacterial RNA polymerase (RNAP) occurs at sequences coding for a GC-rich RNA hairpin followed by a U-rich tract. We used single-molecule techniques to investigate the mechanism by which three representative terminators (his, t500, and tR2) destabilize the elongation complex (EC). For his and tR2 terminators, loads exerted to bias translocation did not affect termination efficiency (TE). However, the force-dependent kinetics of release and the force-dependent TE of a mutant imply a forward translocation mechanism for the t500 terminator. Tension on isolated U-tracts induced transcript release in a manner consistent with RNA:DNA hybrid shearing. We deduce that different mechanisms, involving hypertranslocation or shearing, operate at terminators with different U-tracts. Tension applied to RNA at terminators suggests that closure of the final 2-3 hairpin bases destabilizes the hybrid and that competing RNA structures modulate TE. We propose a quantitative, energetic model that predicts the behavior for these terminators and mutant variants.
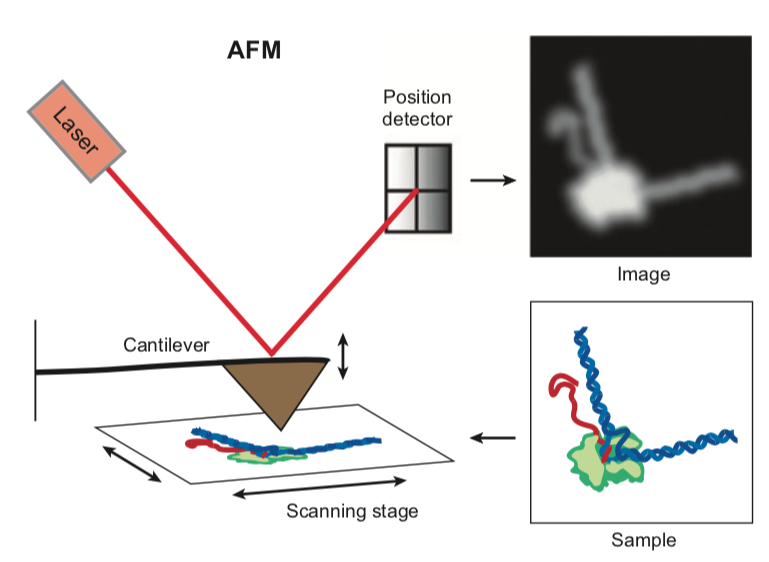
Kristina M. Herbert, William J. Greenleaf, Steven M. Block (2008). “Single-Molecule Studies of RNA Polymerase: Motoring Along” Annual Review Biochemistry
[Review] Single molecule measurements of RNA polymerase: motoring along
Single-molecule techniques have advanced our understanding of transcription by RNA polymerase (RNAP). A new arsenal of approaches, including single-molecule fluorescence, atomic-force microscopy, magnetic tweezers, and optical traps (OTs) have been employed to probe the many facets of the transcription cycle. These approaches supply fresh insights into the means by which RNAP identifies a promoter, initiates transcription, translocates and pauses along the DNA template, proofreads errors, and ultimately terminates transcription. Results from single-molecule experiments complement the knowledge gained from biochemical and genetic assays by facilitating the observation of states that are otherwise obscured by ensemble averaging, such as those resulting from heterogeneity in molecular structure, elongation rate, or pause propensity. Most studies to date have been performed with bacterial RNAP, but work is also being carried out with eukaryotic polymerase (Pol II) and single-subunit polymerases from bacteriophages. We discuss recent progress achieved by single-molecule studies, highlighting some of the unresolved questions and ongoing debates.
William J. Greenleaf, Michael T. Woodside, Steven M. Block (2007) “High-Resolution, Single-Molecule Measurements of Biomolecular Motion” Annual Review Biochemistry
[Review] High-resolution, single-molecule measurements of biomolecular motion
Many biologically important macromolecules undergo motions that are essential to their function. Biophysical techniques can now resolve the motions of single molecules down to the nanometer scale or even below, providing new insights into the mechanisms that drive molecular movements. This review outlines the principal approaches that have been used for high-resolution measurements of single-molecule motion, including centroid tracking, fluorescence resonance energy transfer, magnetic tweezers, atomic force microscopy, and optical traps. For each technique, the principles of operation are outlined, the capabilities and typical applications are examined, and various practical issues for implementation are considered. Extensions to these methods are also discussed, with an eye toward future application to outstanding biological problems.
Greenleaf WJ, and Block SM. (2006) “Single-molecule, motion-based DNA sequencing using RNA polymerase.” Science 313:801.
Single-molecule, motion-based DNA sequencing using RNA polymerase
We present a method for sequencing DNA that relies on the motion of single RNA polymerase molecules. When a given nucleotide species limits the rate of transcription, polymerase molecules pause at positions corresponding to the rare base. An ultrastable optical trapping apparatus capable of base pair resolution was used to monitor transcription under limiting amounts of each of the four nucleotide species. From the aligned patterns of pauses recorded from as few as four molecules, we determined the DNA sequence. This proof of principle demonstrates that the motion of a processive nucleic acid enzyme may be used to extract sequence information directly from DNA.
Abbondanzieri EA*, Greenleaf WJ*, Shaevitz JW, Landick R, Block, SM (2005) “Direct observation of basepair stepping by RNA polymerase.” Nature 438(7067):460-465
Direct observation of basepair stepping by RNA polymerase
During transcription, RNA polymerase (RNAP) moves processively along a DNA template, creating a complementary RNA. Here we present the development of an ultra-stable optical trapping system with ångström-level resolution, which we used to monitor transcriptional elongation by single molecules of Escherichia coli RNAP. Records showed discrete steps averaging 3.7 +/- 0.6 A, a distance equivalent to the mean rise per base found in B-DNA. By combining our results with quantitative gel analysis, we conclude that RNAP advances along DNA by a single base pair per nucleotide addition to the nascent RNA. We also determined the force-velocity relationship for transcription at both saturating and sub-saturating nucleotide concentrations; fits to these data returned a characteristic distance parameter equivalent to one base pair. Global fits were inconsistent with a model for movement incorporating a power stroke tightly coupled to pyrophosphate release, but consistent with a brownian ratchet model incorporating a secondary NTP binding site.
William J. Greenleaf*, Michael T. Woodside*, Elio A. Abbondanzieri, Steven M. Block (2005) “Passive All-Optical Force Clamp for High-Resolution Laser Trapping” Physical Review Letters
Passive all-optical force clamp for high resolution laser trapping
Optical traps are useful for studying the effects of forces on single molecules. Feedback-based force clamps are often used to maintain a constant load, but the response time of the feedback limits bandwidth and can introduce instability. We developed a novel force clamp that operates without feedback, taking advantage of the anharmonic region of the trapping potential where the differential stiffness vanishes. We demonstrate the utility of such a force clamp by measuring the unfolding of DNA hairpins and the effect of trap stiffness on opening distance and transition rates.
Piotr E. Marszalek, William J. Greenleaf, Hongbin Li, Andres F. Oberhauser, Julio M. Fernandez (2000) “Atomic force microscopy captures quantized plastic deformation in gold nanowires” PNAS
Atomic force microscopy captures quantized plastic deformation in gold nanowires
Scanning probe microscopy has become a powerful tool to detect structural changes in small clusters of atoms. Herein, we use an atomic force microscope to measure the length of gold nanowire structures during extension and compression cycles. We have found that nanowires elongate under force in quantized steps of up to three integer multiples of 1.76 A and that they shorten spontaneously in steps of 1.52 A. Our results can be explained by the sliding of crystal planes within the gold nanowires creating stacking faults that change the local structure from face-centered cubic to hexagonal close packed. Our data also show that there can be up to three simultaneous slip events, in good agreement with the tetrahedral arrangement of slip planes in a gold crystal. These experiments provide direct evidence for the mechanism underlying the plastic deformation of a nanowire. A similar approach can be used to examine the atomic events underlying the plastic failure of other metals and their alloys.
William J. Greenleaf, Mark E. Bolander, Gobinda Sarkar, Mary B. Goldring, and James F. Greenleaf. (1998) “Artifical cavitation nuclei significantly enhance acoustically induced cell transfection” World Federation for Ultrasound in Medicine & Biology
Artificial cavitation nuclei significantly enhance acoustically induced cell transfection
The efficiency of ultrasound-mediated gene transfection was enhanced three- to fourfold, compared to previous results, through the use of green fluorescent protein reporter gene, cultured immortalized human chondrocytes and artificial cavitation nuclei in the form of Albunex. Cells were exposed to 1.0-MHz ultrasound transmitted through the bottom of six-well culture plates containing immortalized chondrocytes, media, DNA at a concentration of 40 micrograms/mL and Albunex at 50 x 10(6) bubbles/mL. Transfection efficiency increased linearly with ultrasound exposure pressure with a transfection threshold observed at a spatial average peak positive pressure (SAPP) of 0.12 MPa and reaching about 50% of the living cells when exposed to 0.41 MPa SAPP for 20 s. Adding fresh Albunex at 50 x 10(6) bubbles/mL prior to sequential 1-s, 0.32- or 0.41-MPa exposures increased transfection with each exposure, reaching 43% transfection after four exposures. Efficient in vitro and in vivo transfection now appear possible with these enhancements.